
Rossmeisl Group - Theoretical Catalysis
In our group we work with theoretical catalysis. Which is based on atomic scale simulations and dynamics. In this way we have a direct way into the structure, reactions and intermediates at an atomic level.
Professor Jan Rossmeisl is center leader of the Center of Excellence Center for High Entropy Alloy Catalysis (CHEAC).
Our main research fields are:
• Electrocatalysis for various reactions: water splitting, CO2 reduction ect.
• Screening of semi conductor materials
The hypothesis is that when understanding the process on an atomic scale it is possible to improve both the selectivity and minimize the over-potential (energy loss, for electrochemistry) in the reactions. Finally, when materials are found, they will be tested experimentally with our many partners.
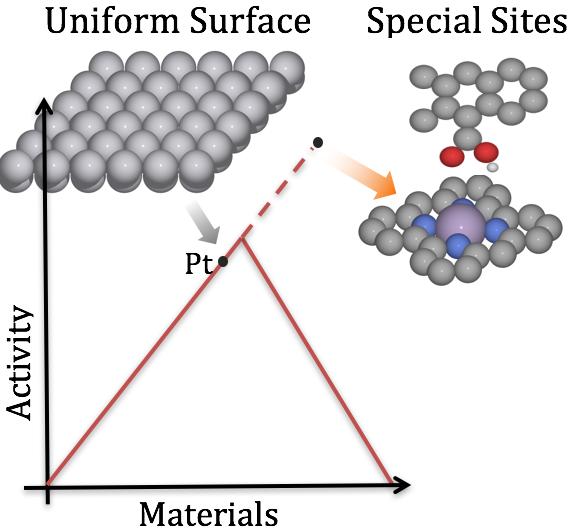
For the computer simulations mainly state of the art periodic Density Functional Theory (DFT) are be applied. DFT simulations offer the right trade-off between accuracy and system size for modeling of catalysis interfaces. The quantum mechanics accurately describe the breaking and formation of chemical bonds and it is possible to model up to ~1000 atoms, which is needed for minimizing the finite size effects. For these calculations we apply our own cluster Katla, at KU, which gives us direct and close by super fast calculations.
KatlaDB - Theoretical Catalysis Database
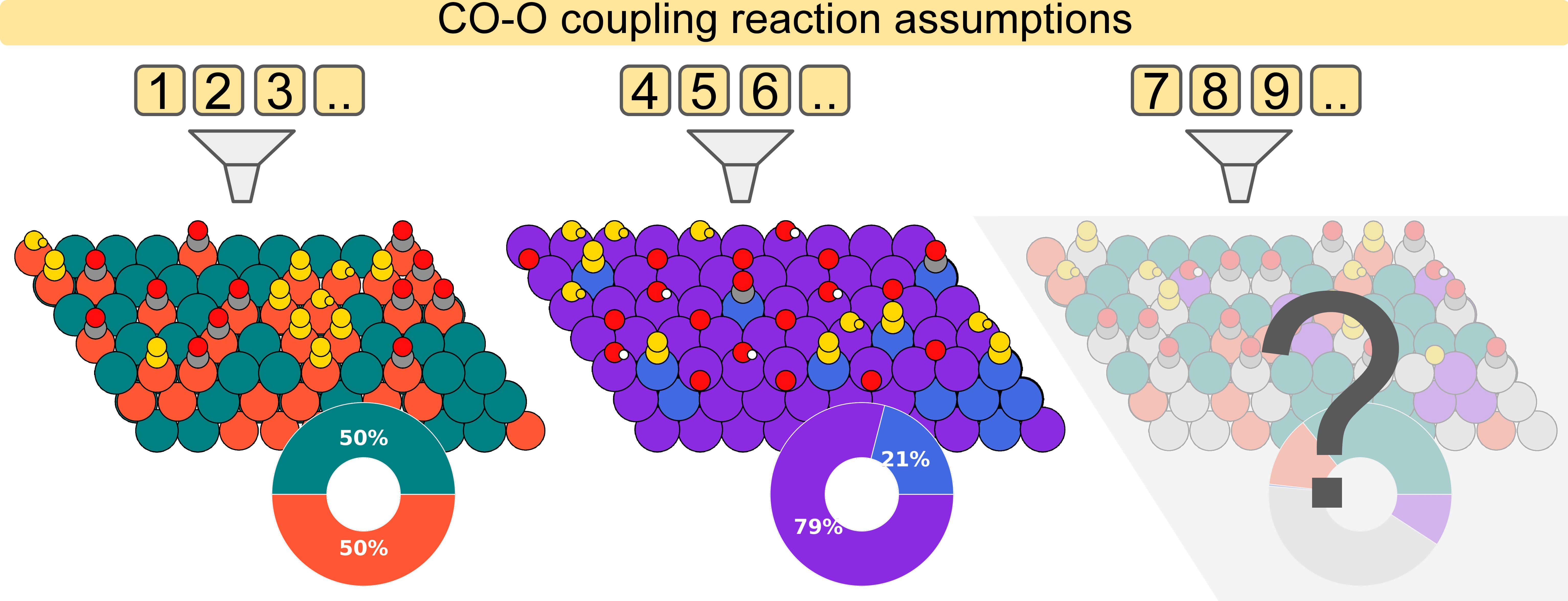 |
Catalytic CO - O Coupling on High-Entropy Alloys: A Composition Optimization Dependent on the Reaction Assumptions
|
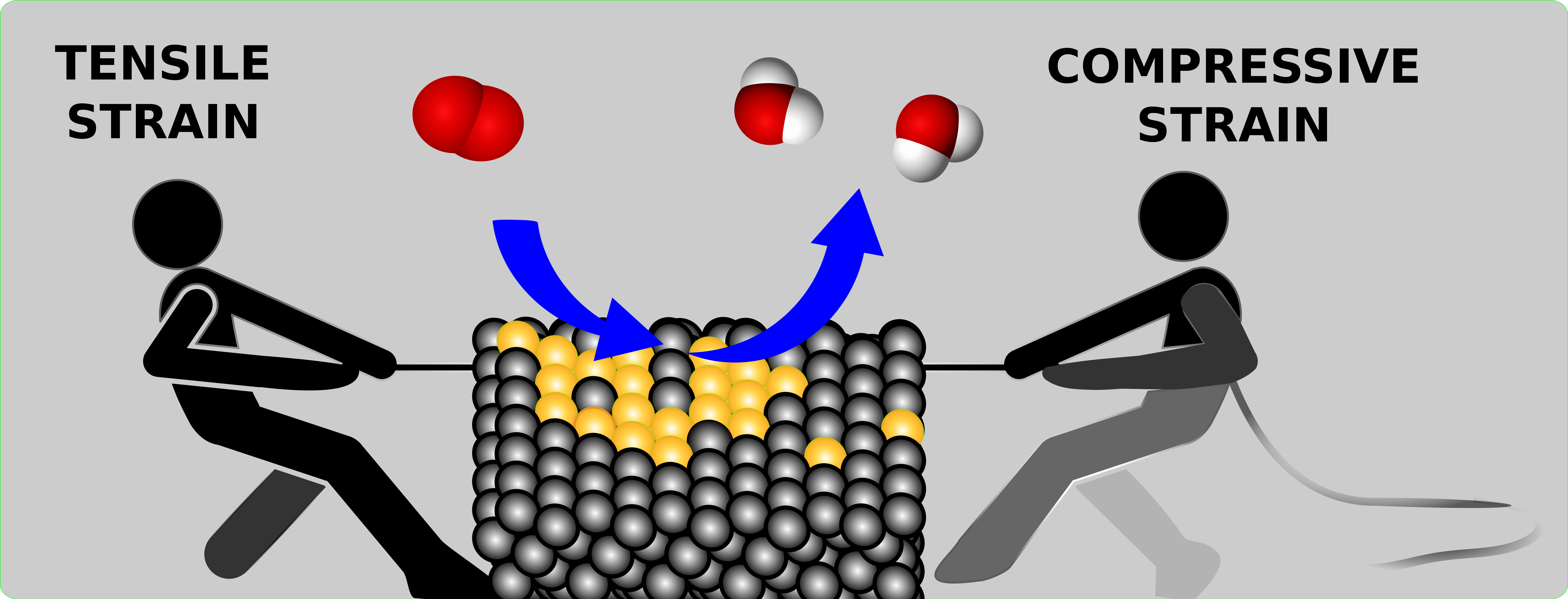 |
Competitive Strain Modulation of Oxygen Reduction Reaction in Monolayer Binary Alloy Surfaces
|
|
Prospects of Using High Entropy Oxides for the Oxygen Evolution Reaction |
|
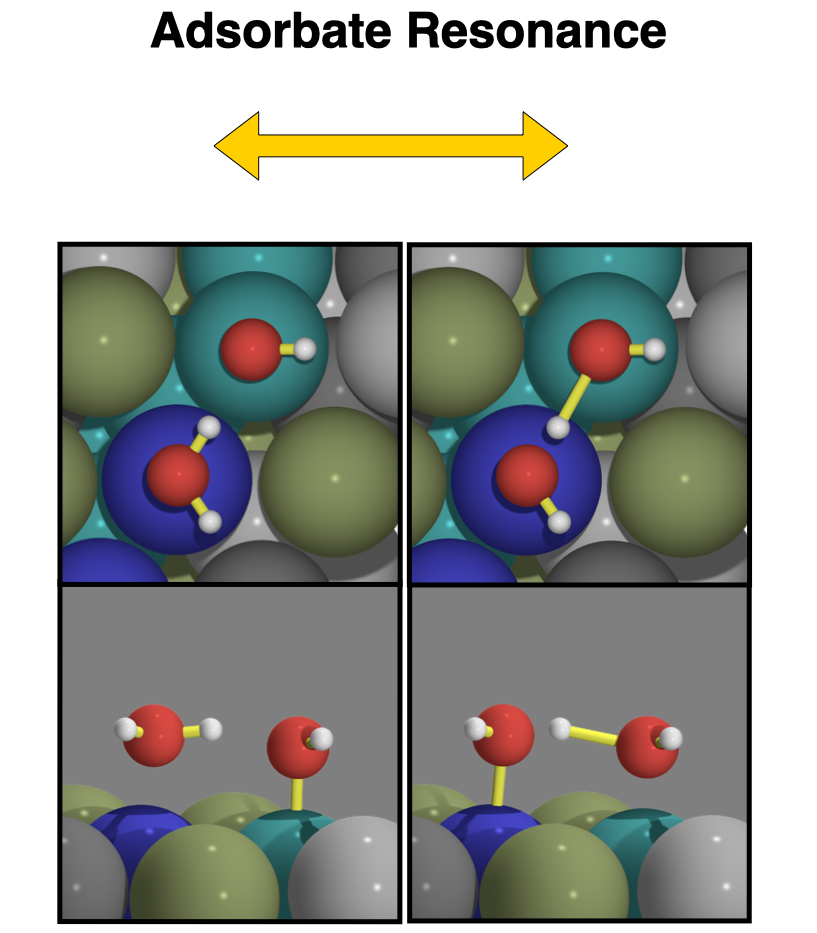 |
Adsorbate Resonance Induces Water-Metal Bonds in Electrochemical Interfaces |
|
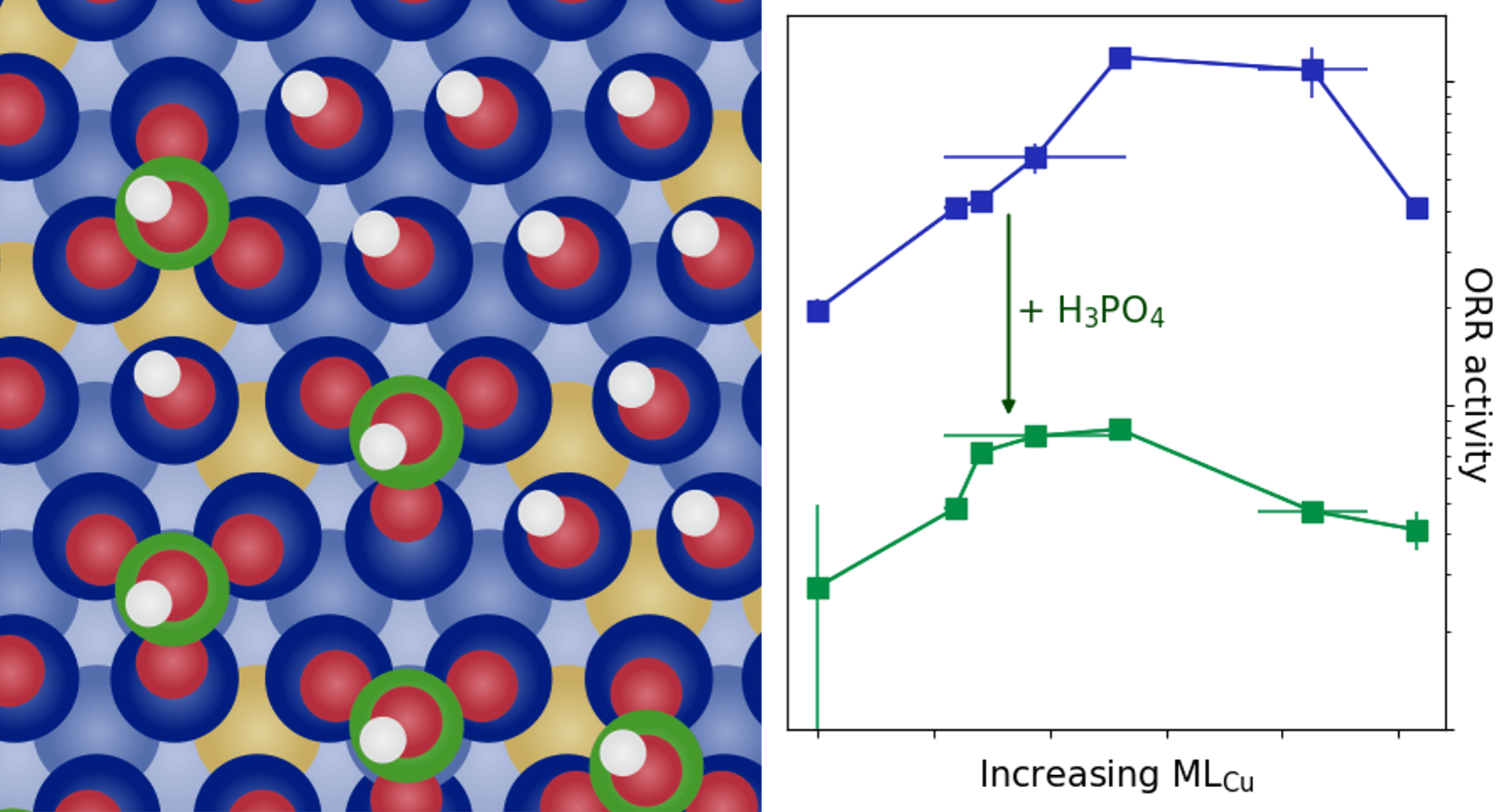
Self-Induced Long-Range Surface Strain Improves Oxygen Reduction Reaction
|
|
 |
Coverage, Repulsion, and Reactivity of Hydrogen on High-Entropy Alloys |
| N2 dissociation on AuCoFeMoRu high-entropy alloys: Circumventing scaling relations and step dependencies
|
|
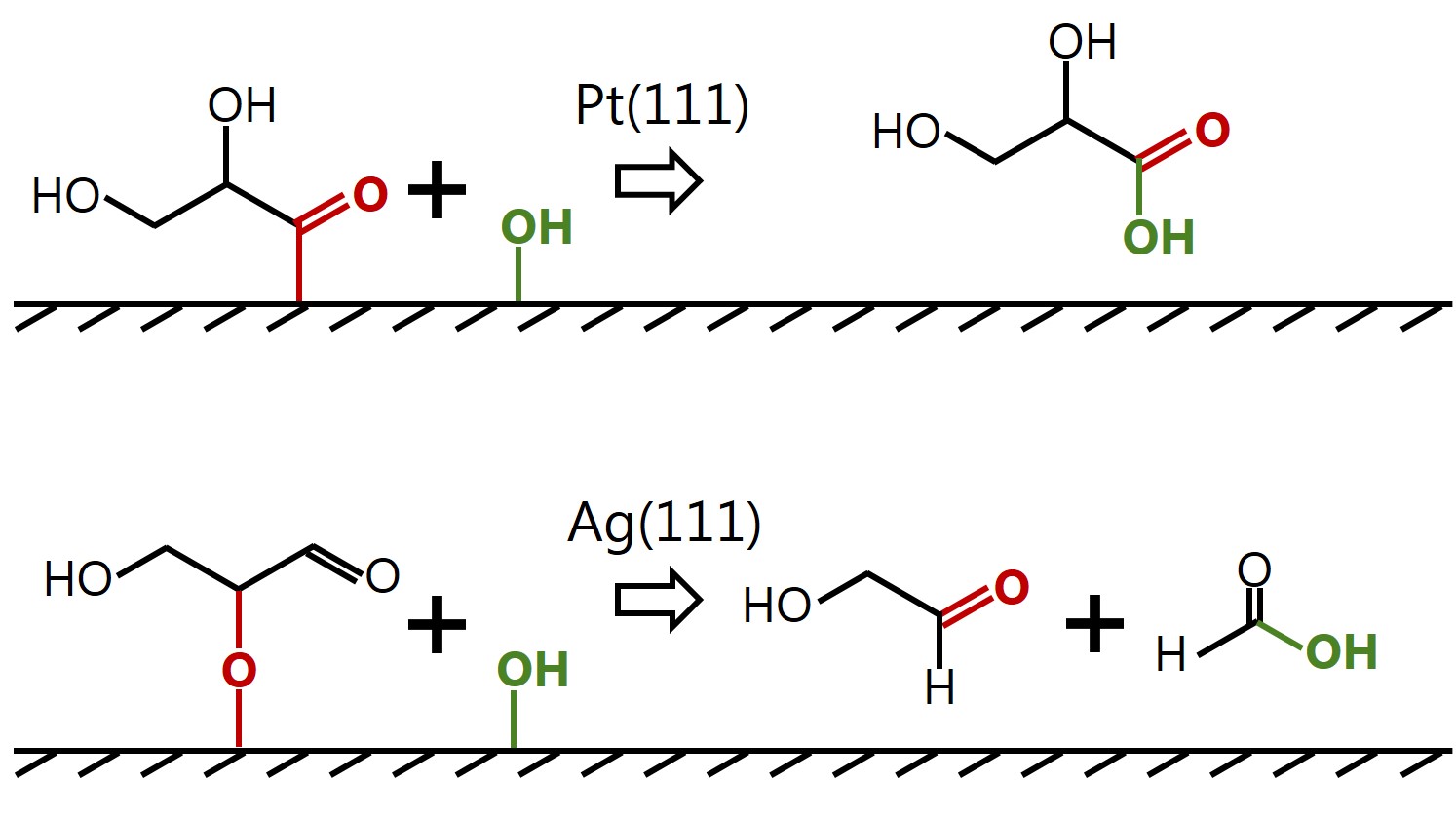 |
Insight into selectivity differences of glycerol electro-oxidation on Pt(111) and Ag(111) |
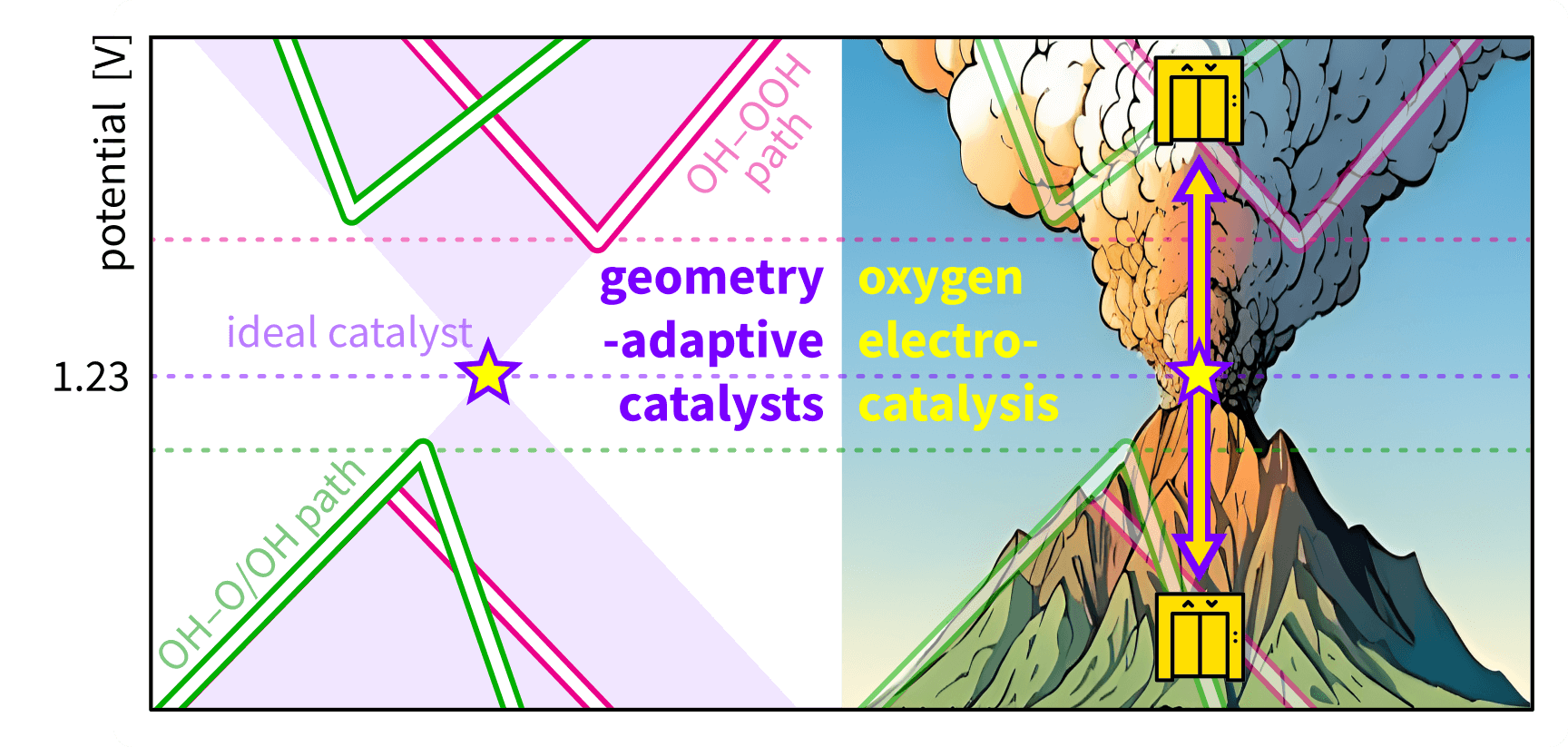 |
Geometry-adaptive catalysts for elevating towards the ideal oxygen electrocatalysis |
 |
|
|
Lattice oxygen evolution in rutile Ru1-xNixO2 electrocatalysts |
|
|
Ru rich Ru-Mn-O phases for selective suppression of chlorine evolution in sea water electrolysis |
|
|
|
|
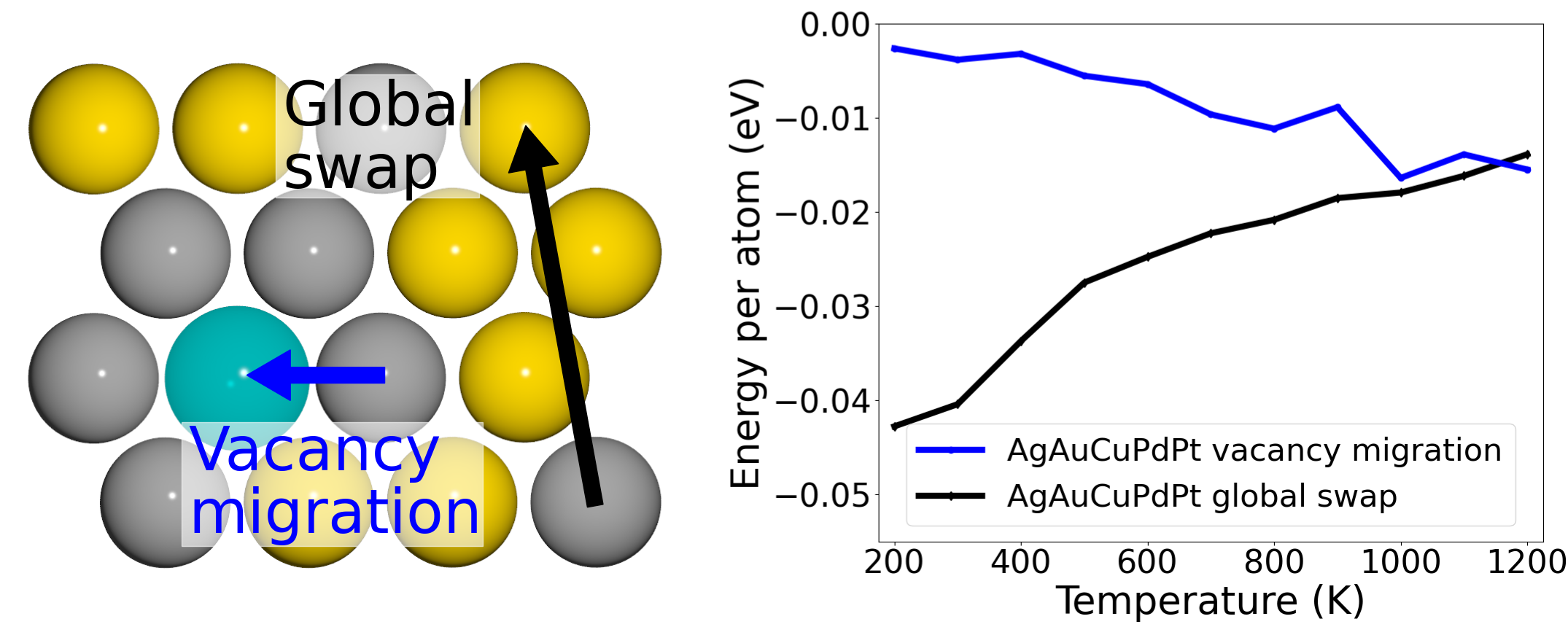 |
Role of vacancies in structural thermalization of binary and high-entropy alloys |
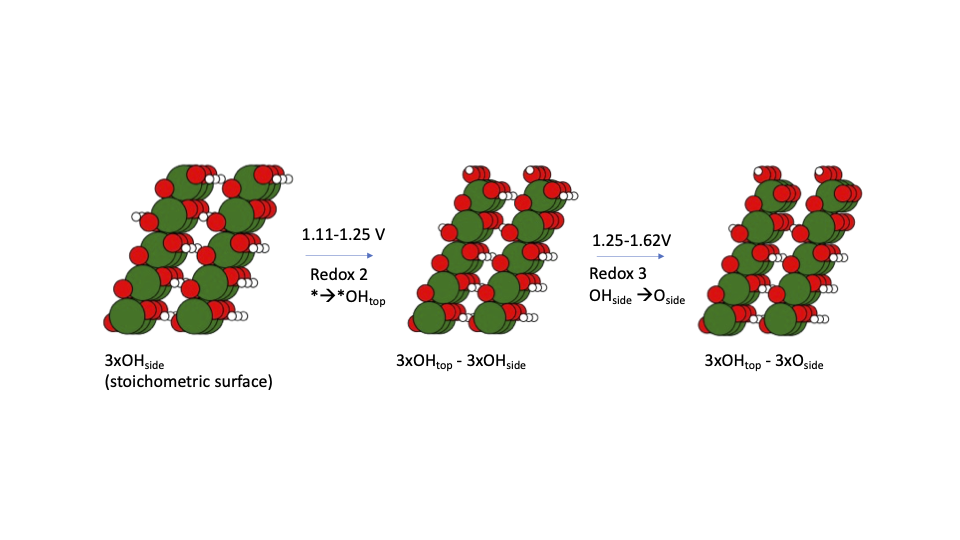 |
Cooperative effects drive water oxidation catalysis in cobalt electrocatalysts through the destabilisation of intermediates |
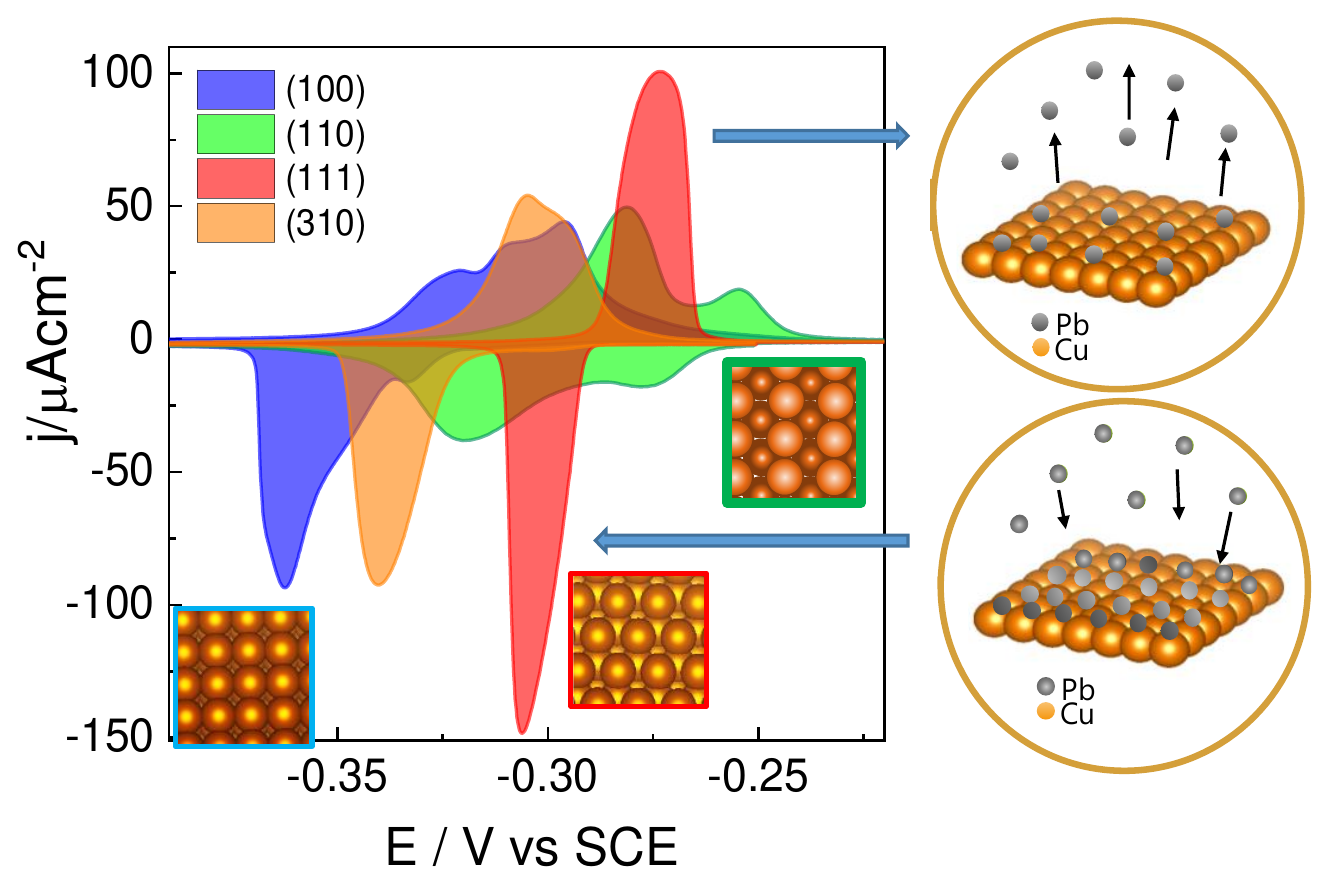 |
Tailoring the facet distribution on copper with chloride |
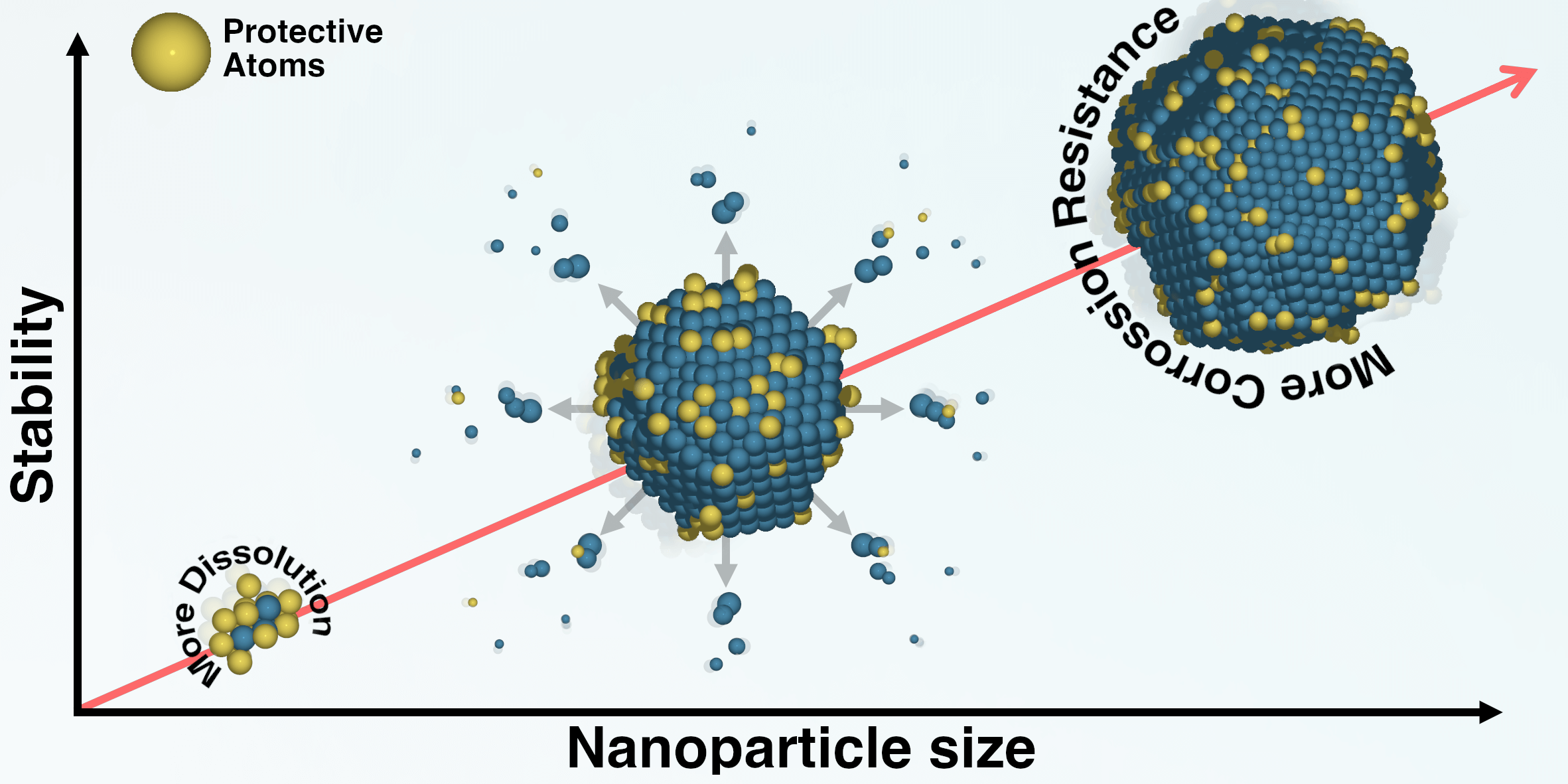 |
Tuning the chemical composition of high-entropy alloys to prevent their dissolution |
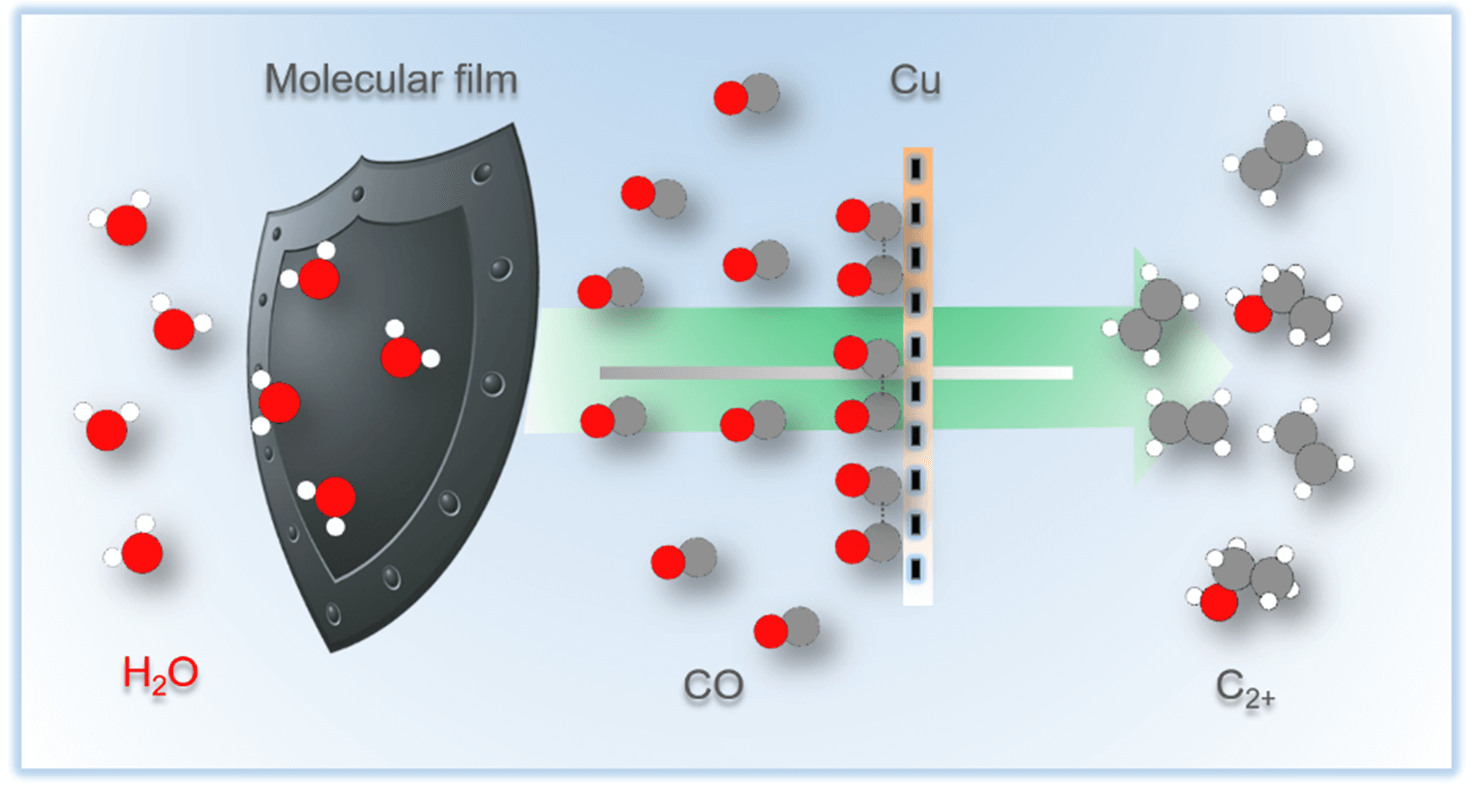 |
Steering carbon dioxide reduction toward C–C coupling using copper electrodes modified with porous molecular films
|
 |
|
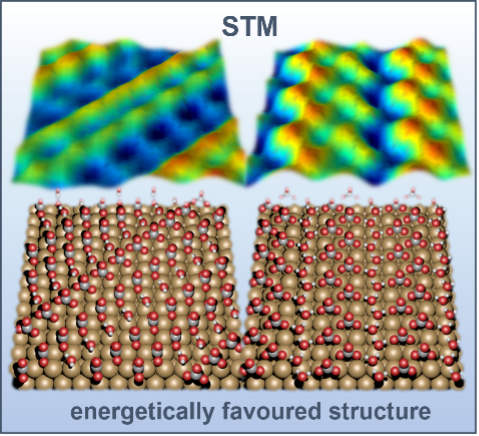
Unravelling the effects of active site densities and energetics on the water oxidation activity of iridium oxides |
|
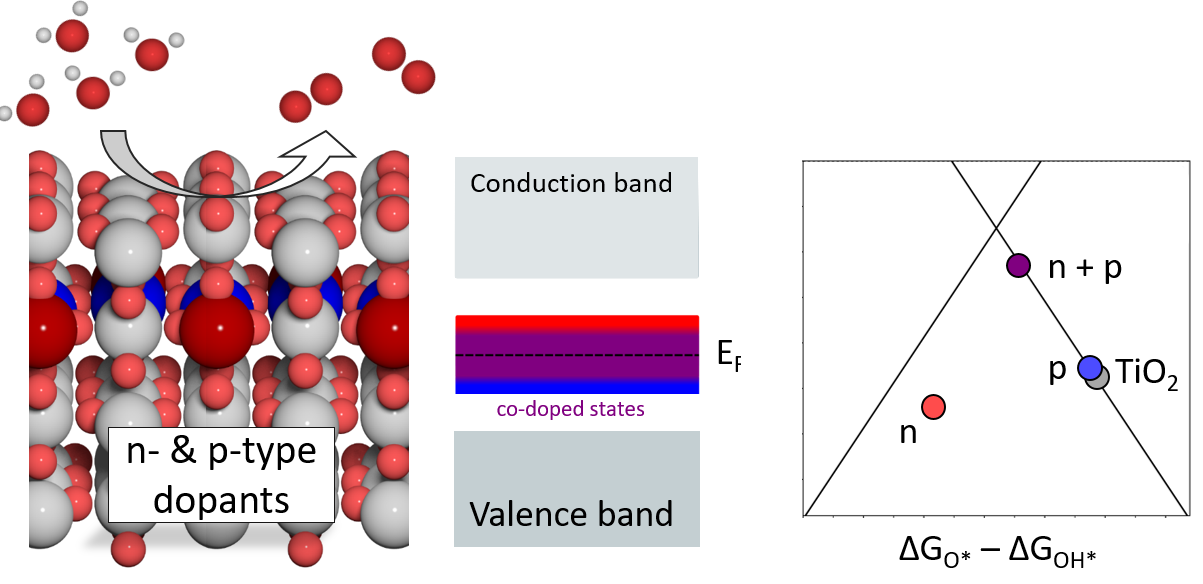 |
Synergistic effect of p-type and n-type dopants in semiconductors for efficient electrocatalytic water splitting
|
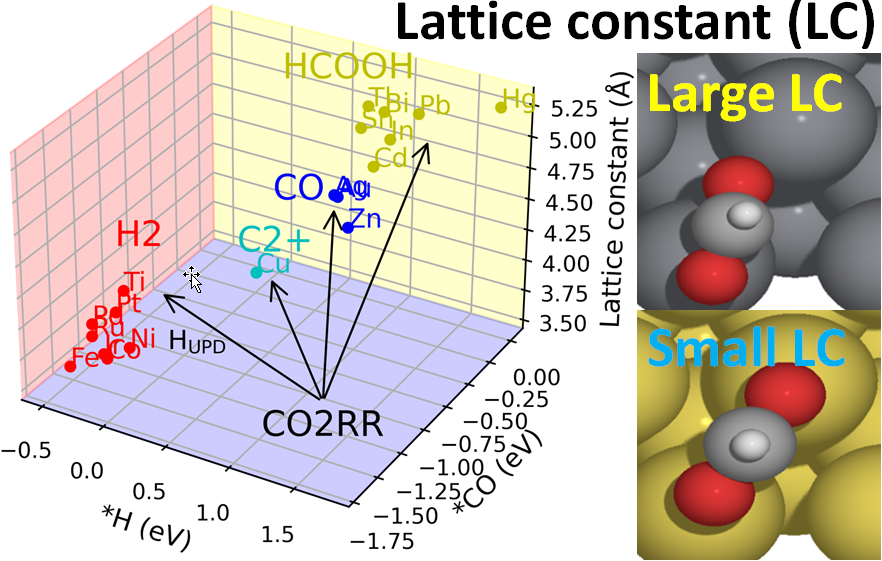
CO2 reduction on Ag-Cu-Pd |
|
 |
Structure of the (bi)carbonate adlayer on Cu(100) electrodes |
Modeling electrochemical proton adsorption at constant potential with explicit charging |
|
Following Paths of Maximum Catalytic Activity in the Composition Space of High-Entropy Alloys |
|
 |
Bridging the Catalyst Reactivity Gap Between Au and Cu for the Reverse Water Gas Shift Reaction |
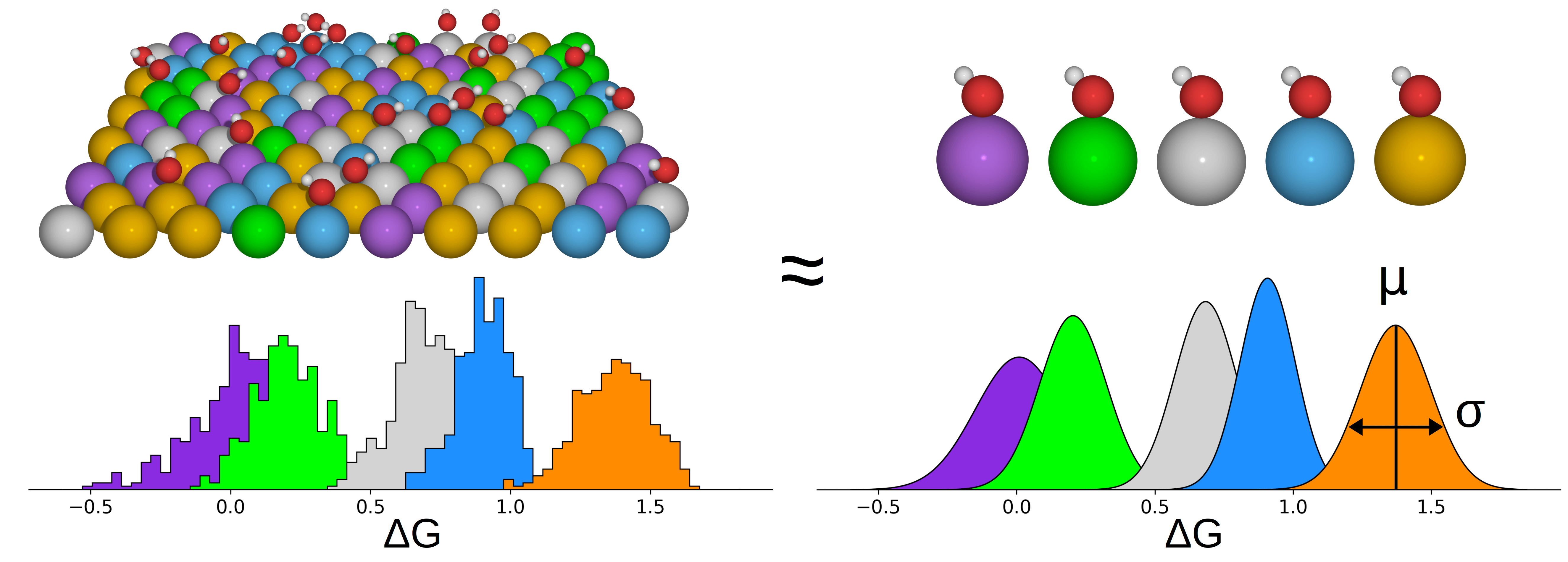 |
A Mean Field Model for Oxygen Reduction Electrocatalytic Activity on High-Entropy Alloys |
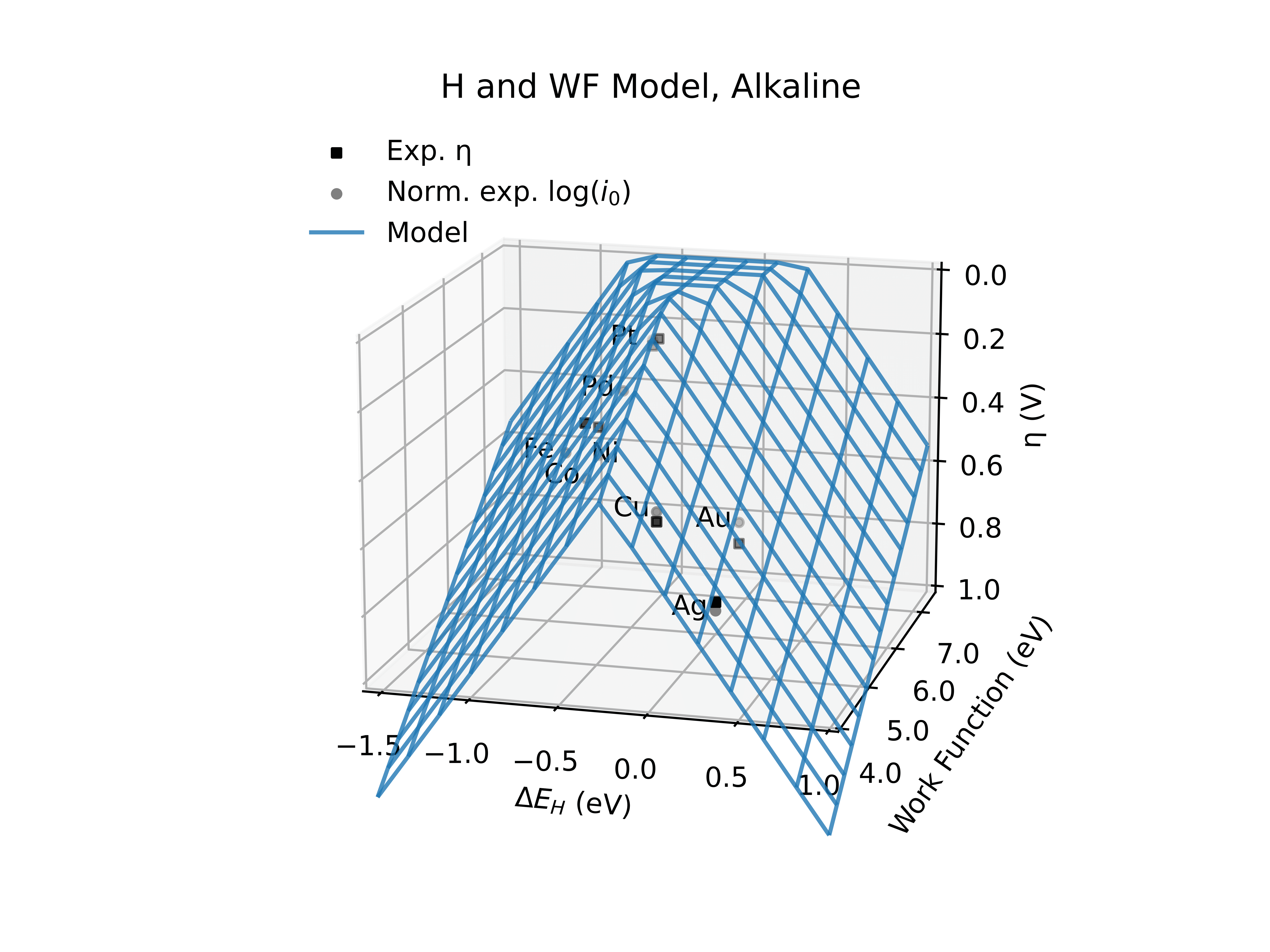 |
Predicting Catalytic Activity in Hydrogen Evolution Reaction |
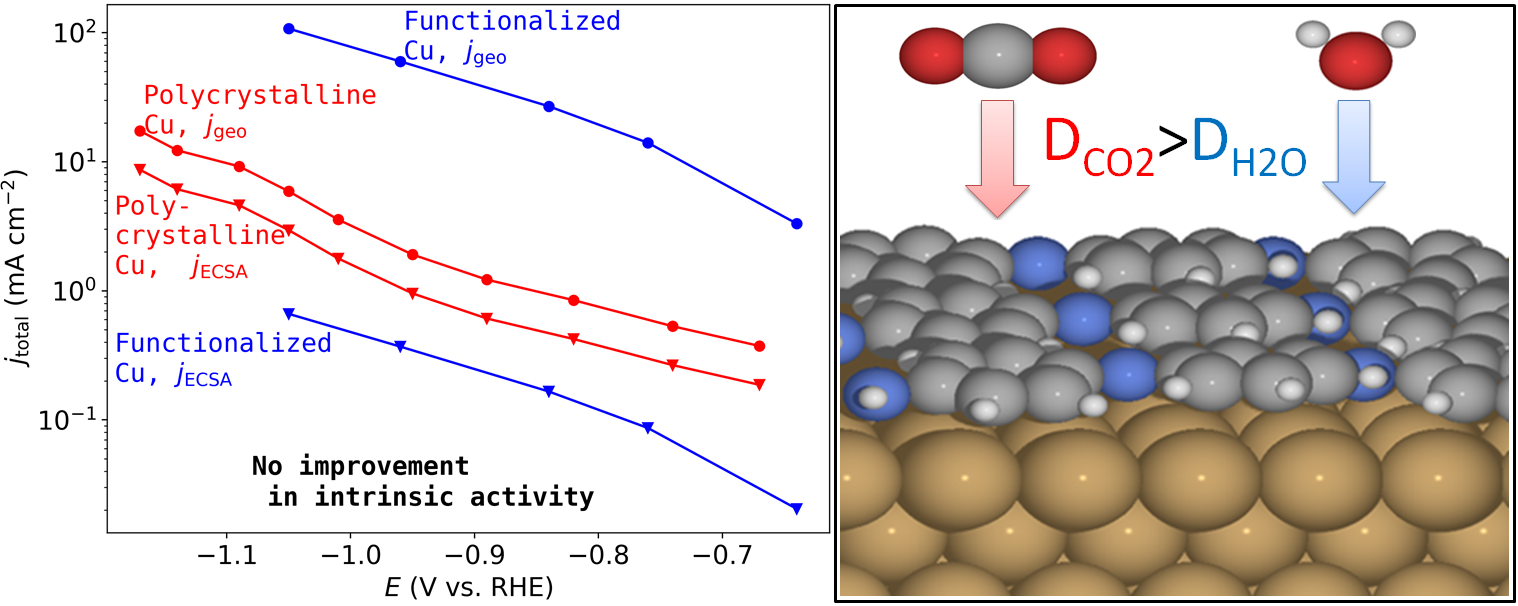 |
Selectivity and Intrinsic Activity of Functionalized Cu Surfaces: Can the CO2RR be Improved on Cu?
|
 |
Formic Acid Oxidation Reaction |
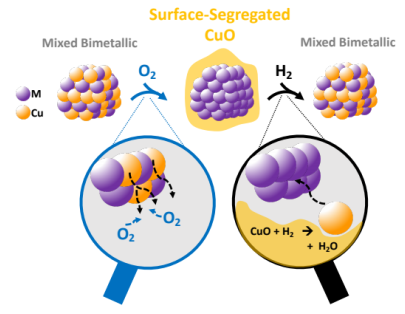 |
Exploring the Mobility of Cu in Bimetallic Nanocrystals to Promote Atomic-Scale Transformations under a Reactive Gas Environment |
High entropy oxides for the oxygen evolution reaction |
|
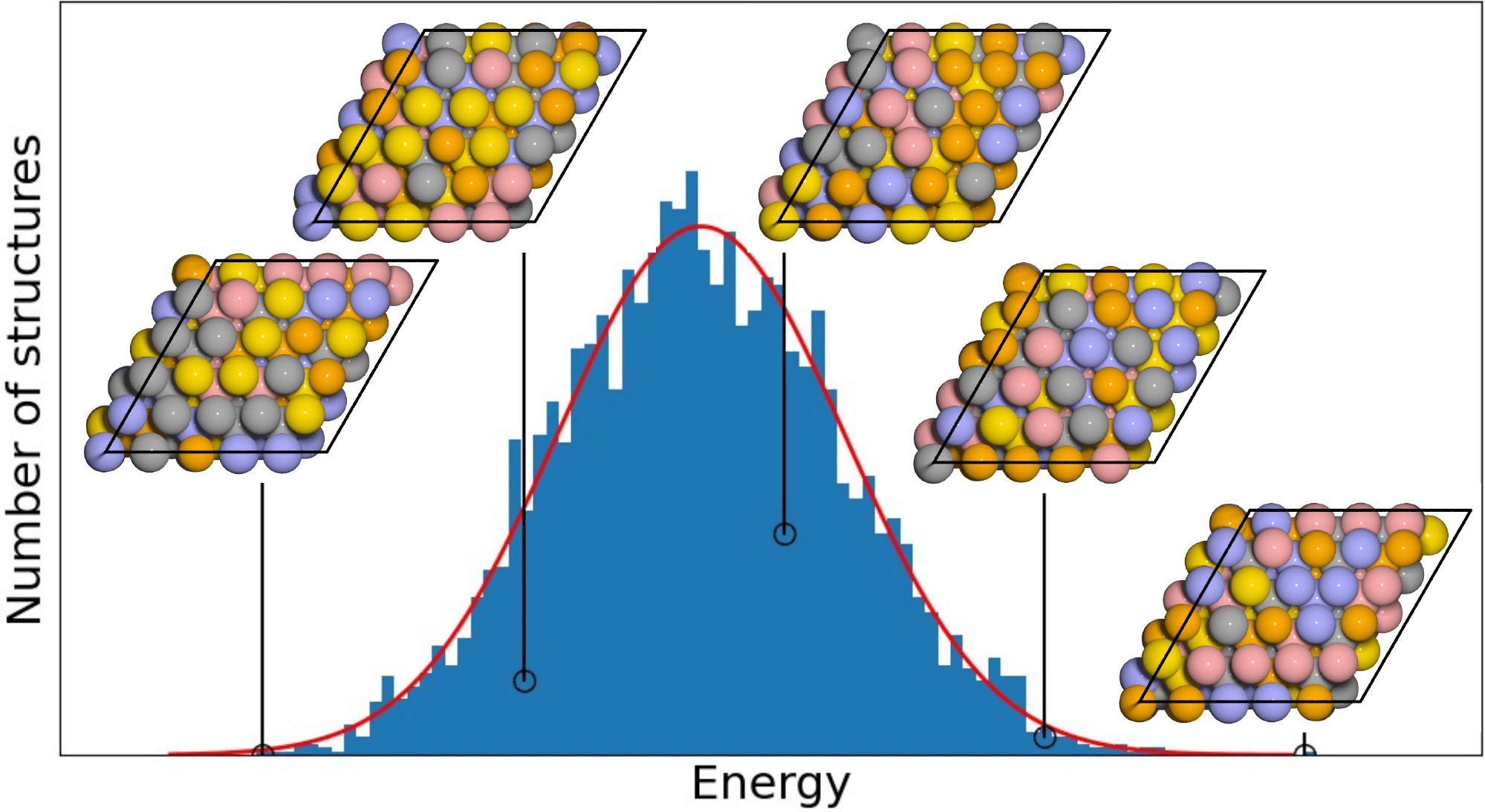 |
Local order in AgAuCuPdPt high entropy alloy surfaces |
|
|
Catalytic CO2/CO Reduction: Gas, Aqueous and Aprotic phase |
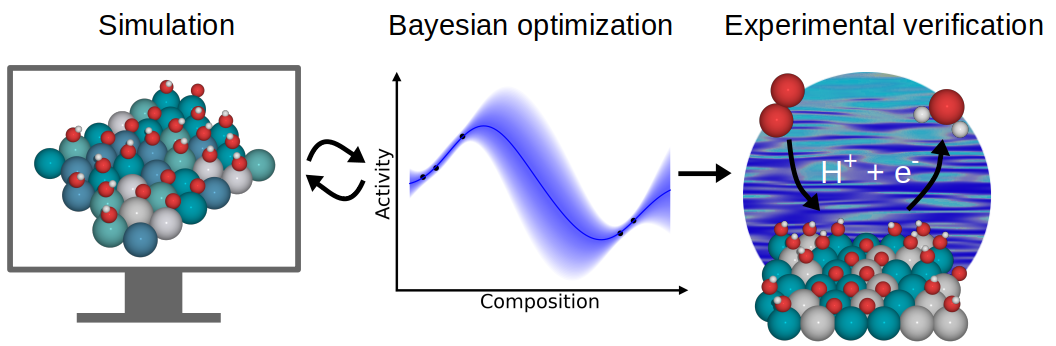 |
|
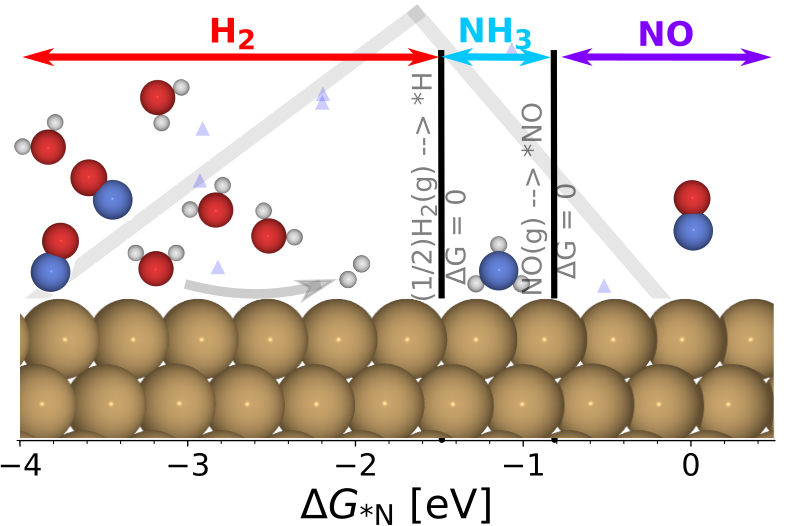 |
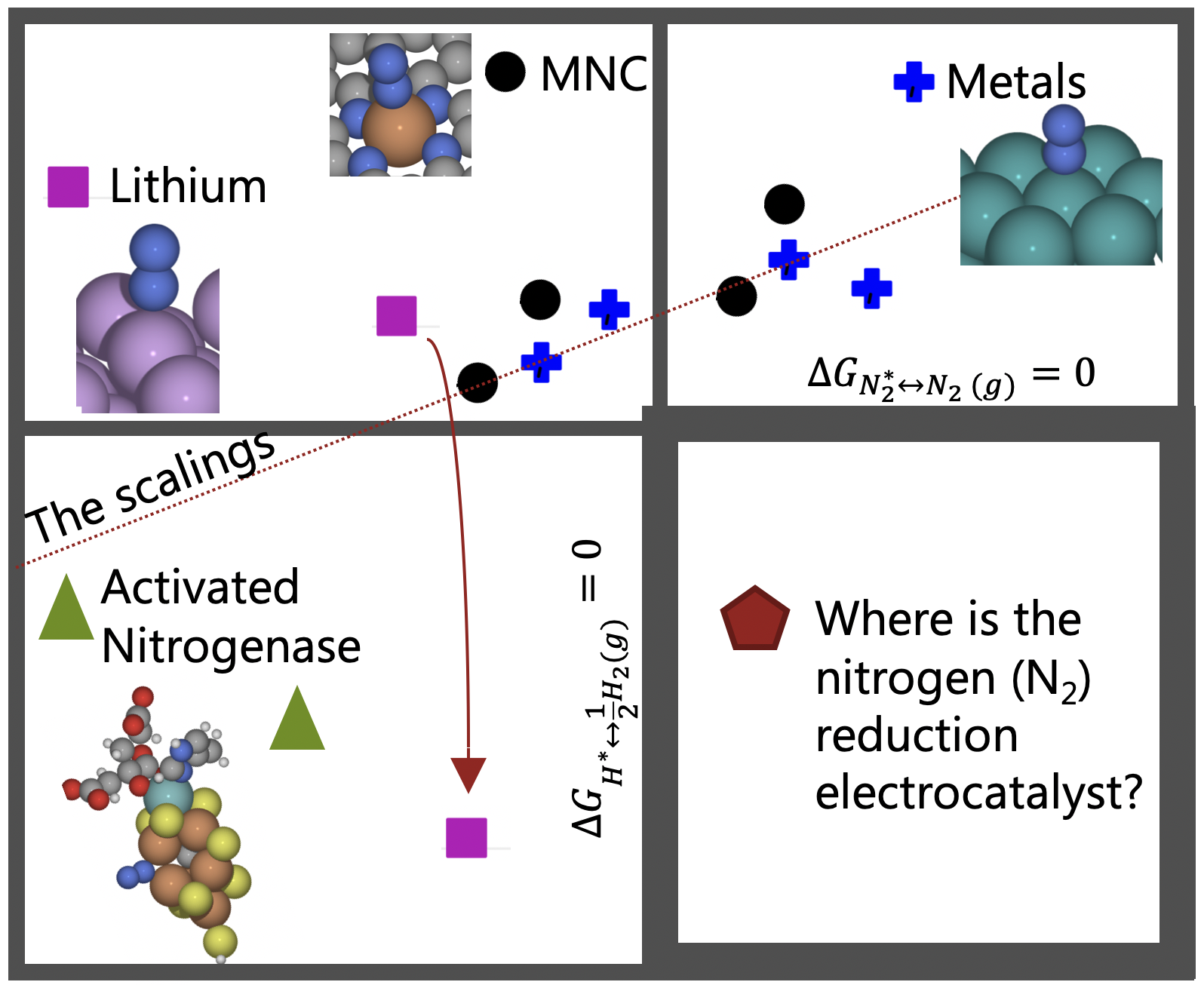
Electrochemical Nitric Oxide Reduction on Metal Surfaces
*Correspondence: jan.rossmeisl@chem.ku.dk DOI: (to be provided) Link to zip file containing data and scripts for reproduction: XX (XX MB) |
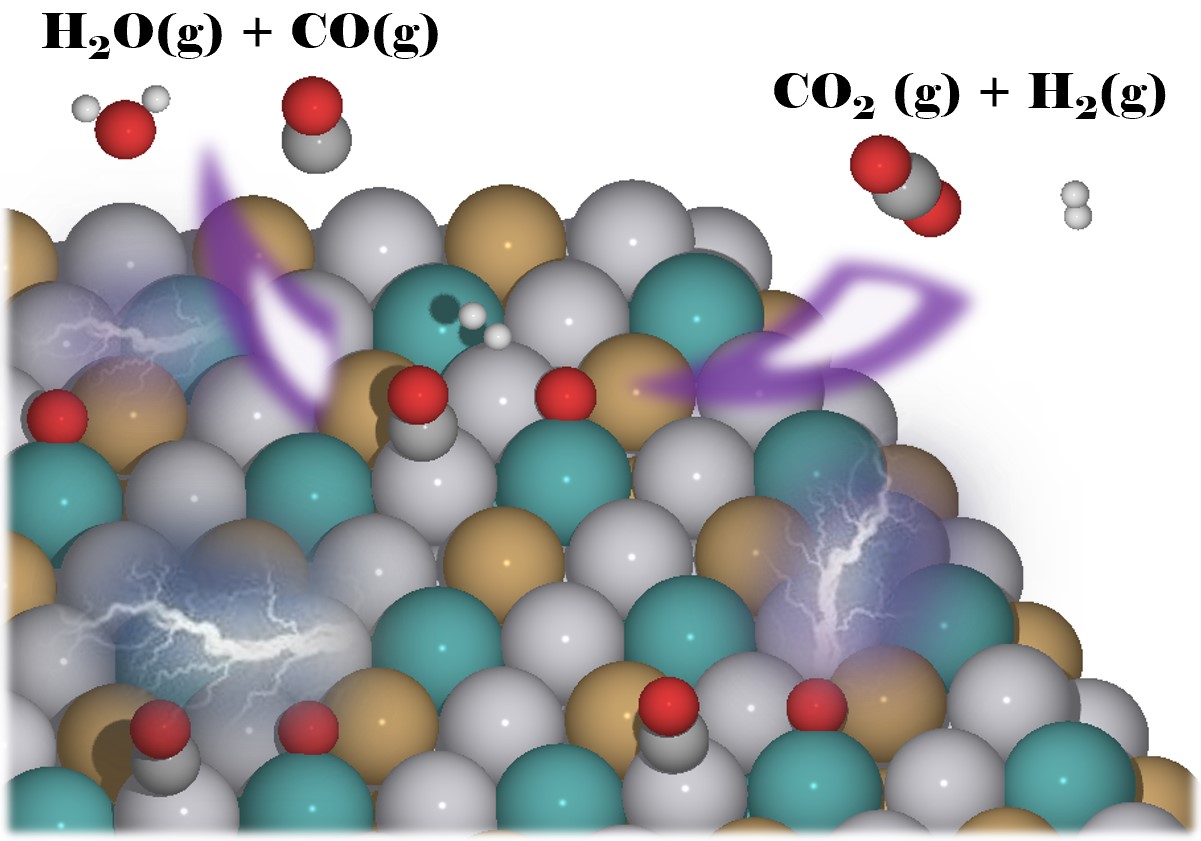 |
Rationally tailoring catalysts for the CO oxidation reaction by using DFT calculationsWe use O on-top and CO on-top adsorption energies to estimate O bridge and O hollow adsorption energies, and predict activation and reaction energies for CO oxidation reactions (COOR). |
 |
|
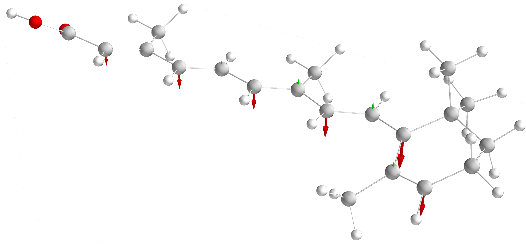 |
We investigate the exchange interaction of deprotonated retinoic acid molecules across hydrogen bonds.
|
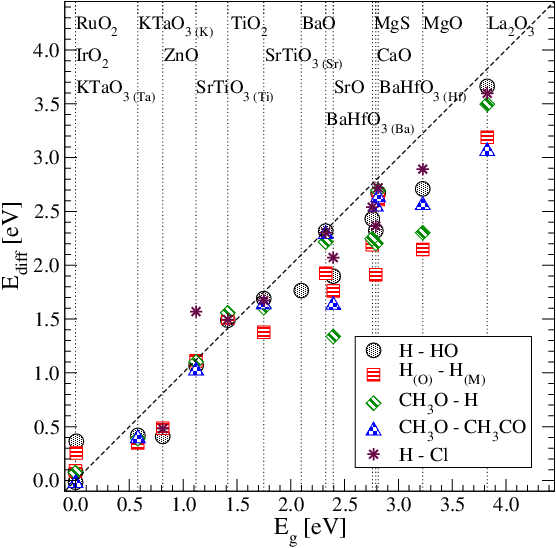 |
We investigate the interaction energy of coadsorbed fragments on band gap oxides and we propose to use the band gap as a descriptor of the interaction energy.
|
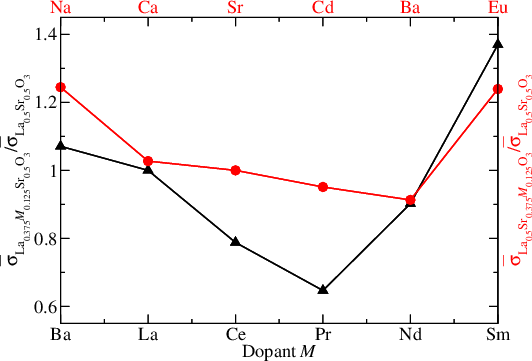 |
We have calculated the electrical conductivity of the SOFC cathode contact material La1−xSrxCoO3−δ at 900 K. Furthermore, we have studied the chemistry of neutral and charged intrinsic and extrinsic defects (dopants) in La0.5Sr0.5CoO3 and calculated the conductivity of the doped systems. In particular, we find that doping with Sm on the La site should enhance the conductivity.
|
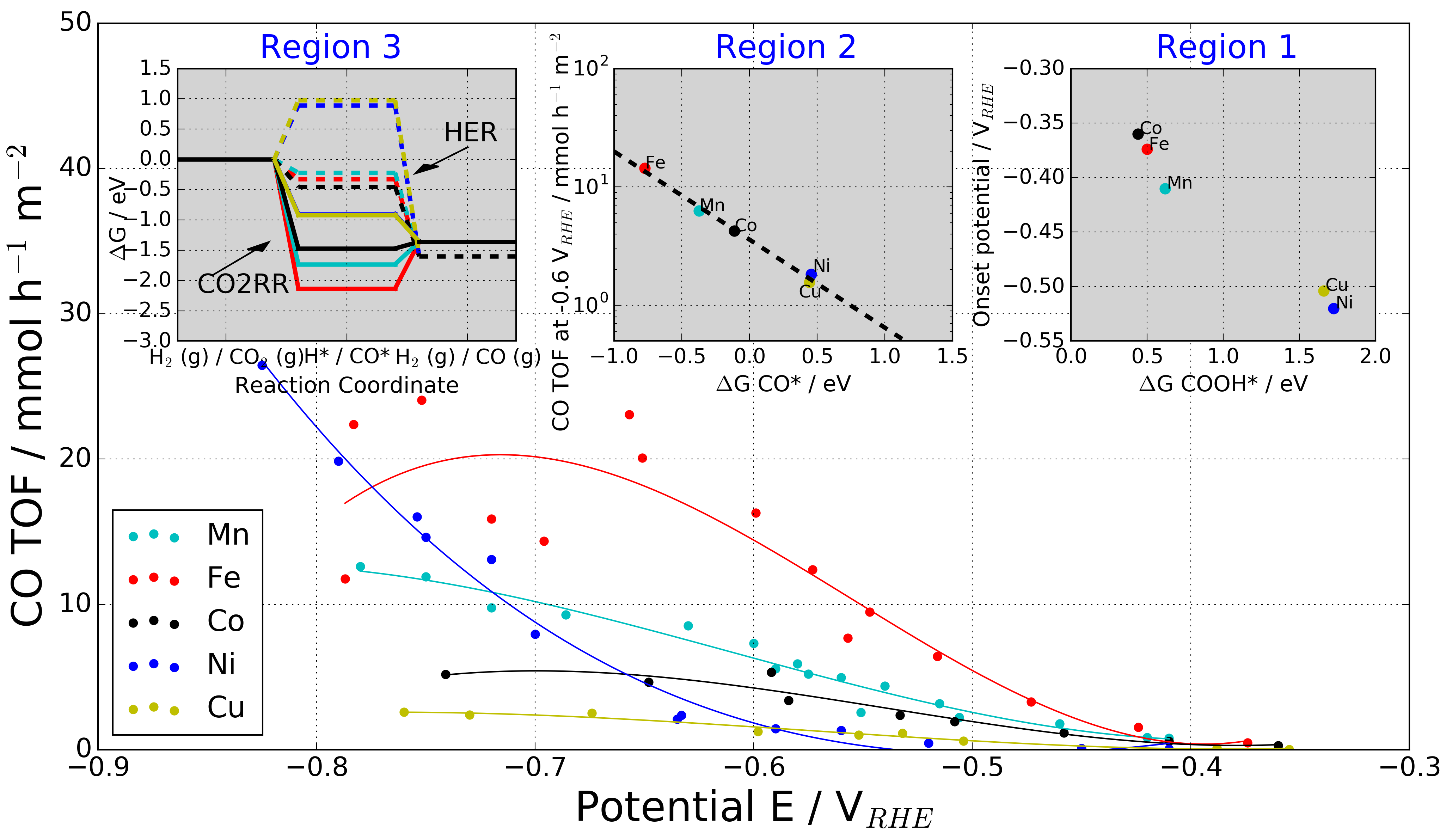
 |
The data is supplementary material for the paper, which includes all structures and binding energies calculated. Further, figures have been plotted with labels for all metals and porphyrine-like structures.
|
|
|
Catalyst design criteria and fundamental limitations in the electrochemical synthesis of dimethyl carbonate We demonstrate how density functional theory (DFT) calculations and experiments advance our understanding of electrocatalytic production of chemicals. Using DFT, we form design criteria for dimethyl carbonate electrosynthesis on metallic surfaces. These criteria allow us to identify copper as an interesting candidate for the electrode material as being selective to dimethyl carbonate while requiring ≈ 1 V lower potential than a gold electrode. Electrode stability issues show that the design criteria presented herein are necessary but not sufficient requirements that the ideal electrode should satisfy.
|
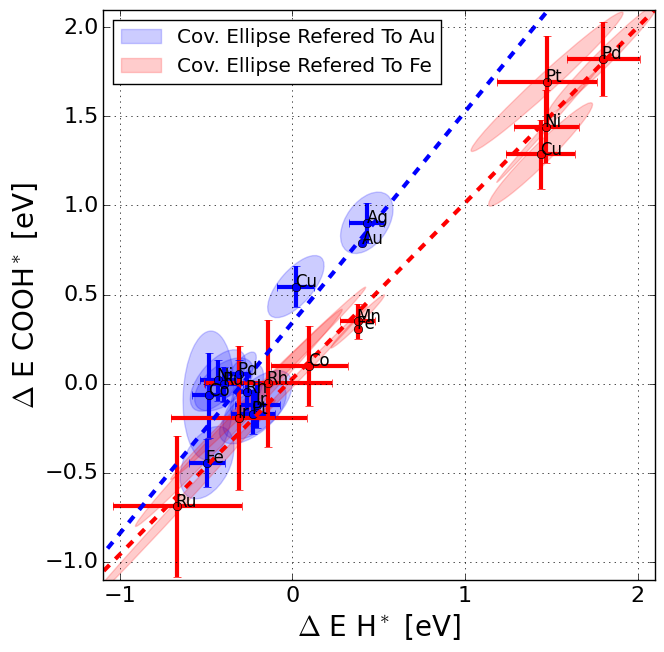
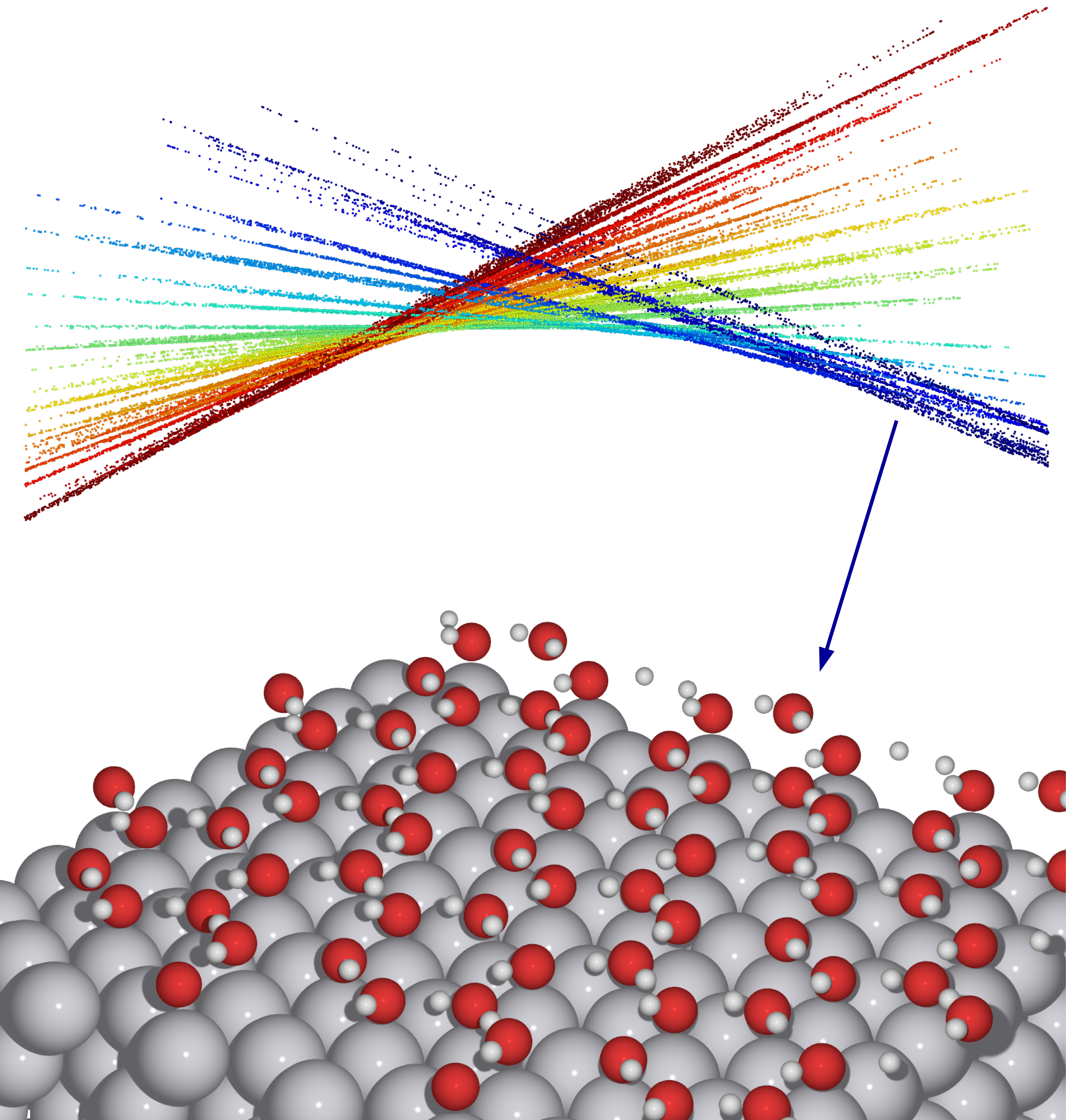 |
We present atomic-scale structures of the Pt(111)/water interface, by calculating distributions of atomic distances as functions of pH. The structure of the Pt(111)/water interface is a particularly interesting model system in electro-catalysis for proton exchange reactions, especially the oxygen reduction reaction in polymer electrolyte membrane fuel cells. Further insight into such reactions requires accurate simulations of the electrolyte structure in the interface. The study displays many interesting details in the behaviour of the electrolyte structure, e.g. that the electrolyte structure average responds to the presence of protons by a H-down water orientation and that hexagonal adsorbed water layers are present only when they are anchored at the surface by HO*. New adsorbate configurations were also found at 5/12 ML coverage of HO*, suggesting an explanation for reported cyclic voltammetry experiments. The present study is a step towards a more complete understanding of the structure of the electrochemical interface on the atomic scale.
|
 |
The data is supplementary material for the paper, which includes all structures, binding energies and energy ensembles. Further, plotting script is given to plot Figure 1 of the paper and the layout can be used to also achieve and plots other data from the database.
|
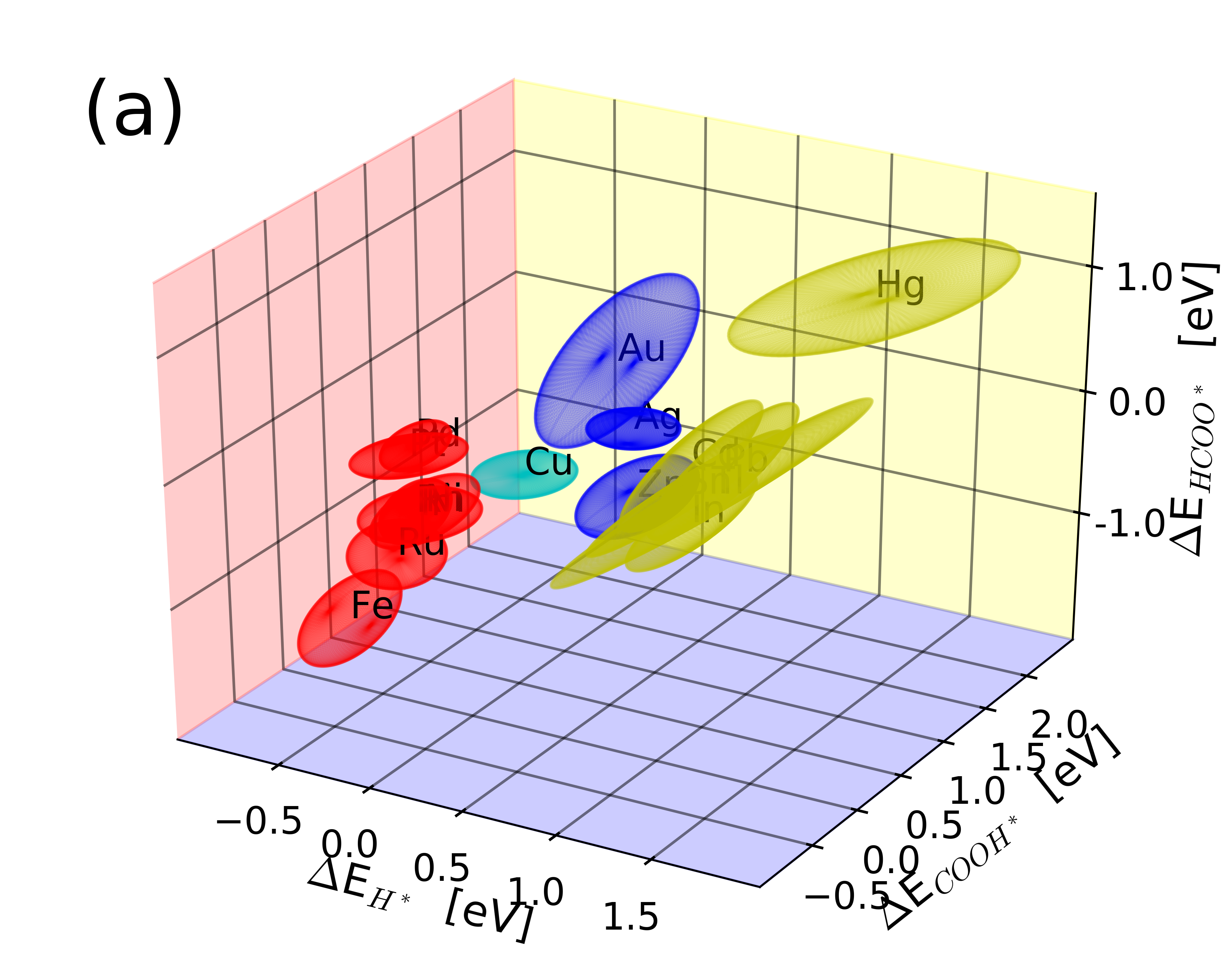
 |
The data is supplementary material for the paper, which includes all structures and binding energies calculated. Further, experimental data together with plotting script for Figure 5 in the paper can be downloaded.
|
|
Enhanced Carbon Dioxide Electroreduction to Carbon Monoxide over
Defect-Rich Plasma-Activated Silver Catalysts The data is supplementary material for the paper, which includes all structures and binding energies calculated.
|
|
|
Accessing the Inaccessible: Analyzing the Oxygen Reduction Reaction in the Diffusion Limit A numerical site-blocking model to simulate ORR currents in the absence of mass transport limitatons. |
|
|
Fundamental limitation of electrocatalytic methane conversion to methanol A database with all structures and scripts for plotting figures. |
|
|
Oxidation of Ethylene Carbonate on Li Metal Oxide Surfaces
Contains a database file with structures and energies, and scrips for plotting all figures from the paper. |
|
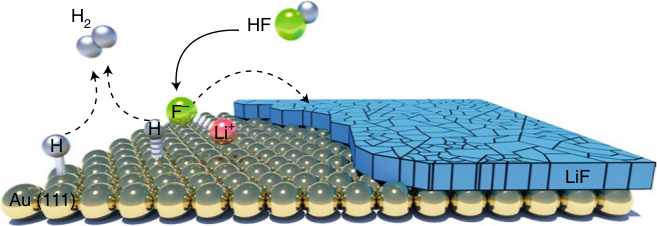 |
Electrocatalytic transformation of HF impurity to H2 and LiF in lithium-ion batteries
We have discussed the formation of H2 and LiF from a dissociation of HF in Li-ion batteries. The database contains all the calculations used in the paper to produce the phase diagram of Li and identify trends. |
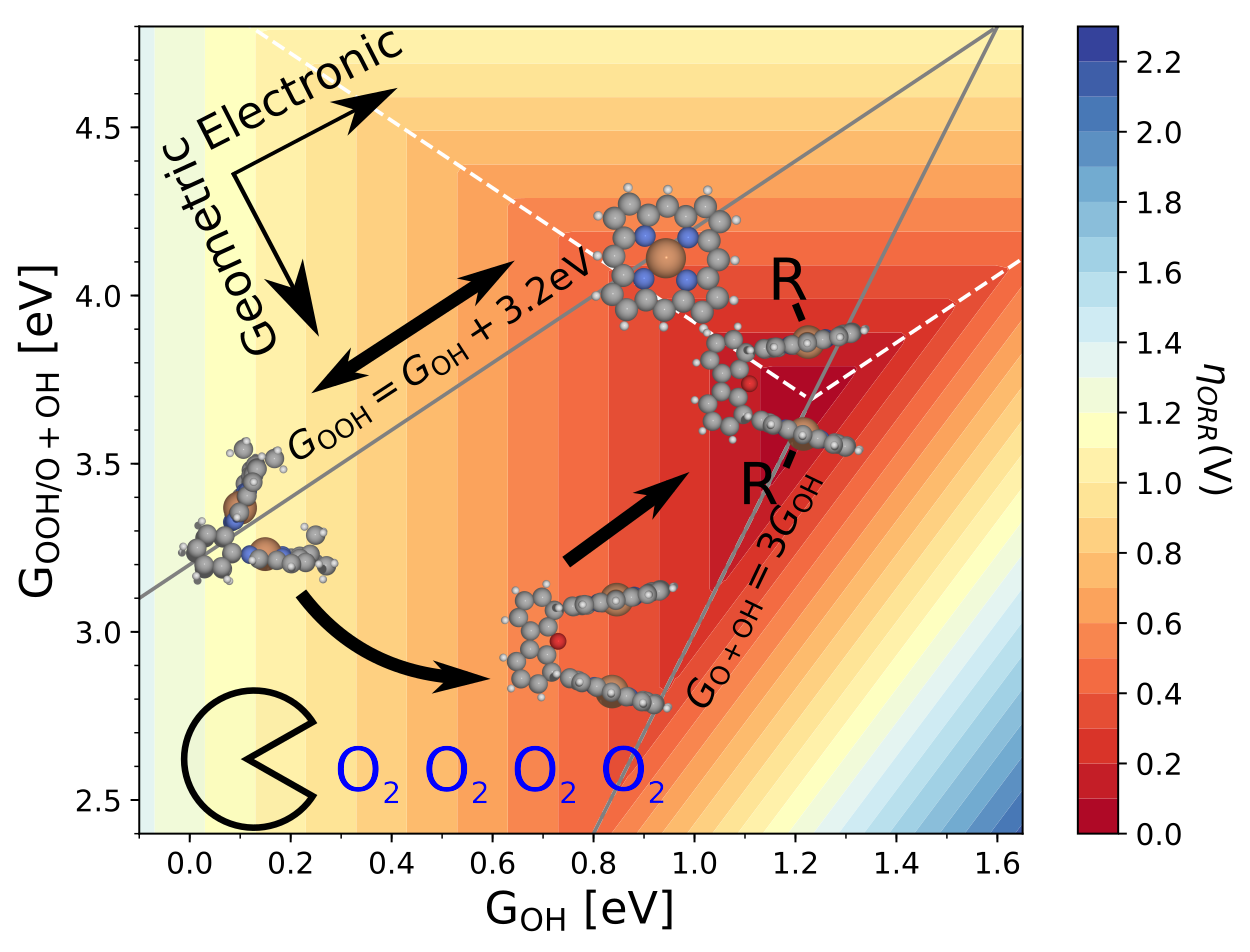 |
Climbing the 3D Volcano for the Oxygen Reduction Reaction Using Porphyrin Motifs
Contains a database file with structures and energies, and scrips for plotting all figures from the paper. |
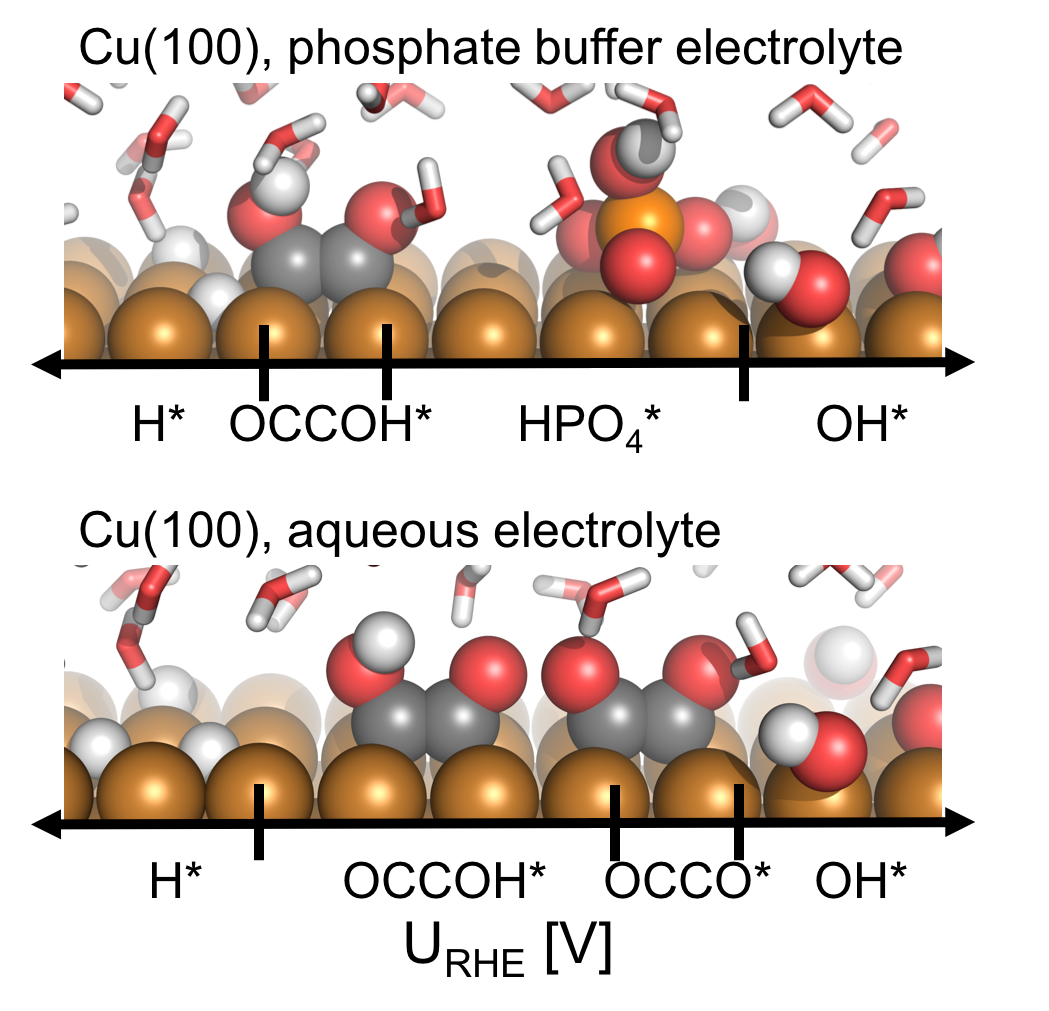 |
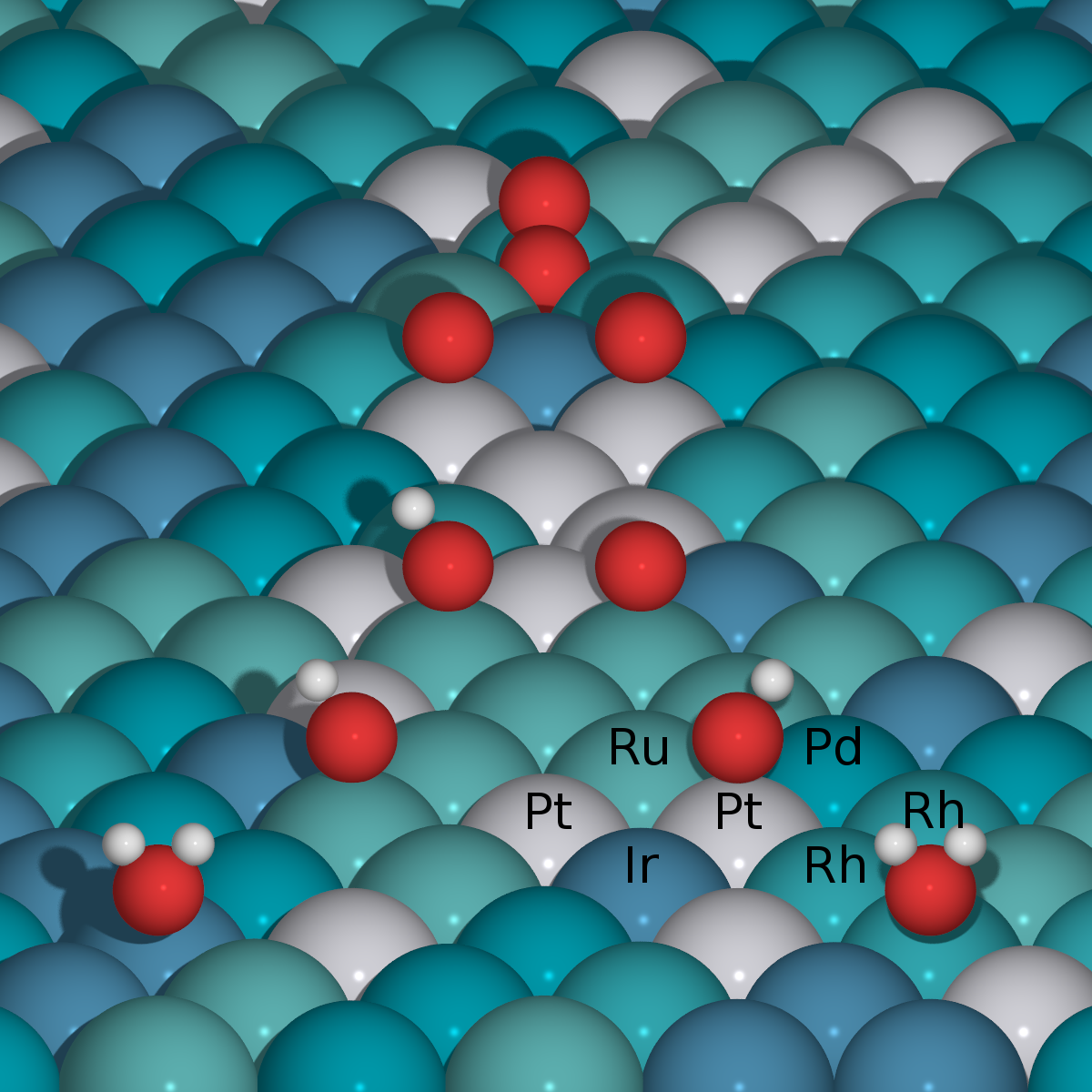
Electrochemical CO reduction: A property of the electrochemical interface
Contains a .zip file with multiple folders with databases and scrips for plotting figures from the paper. |
 |
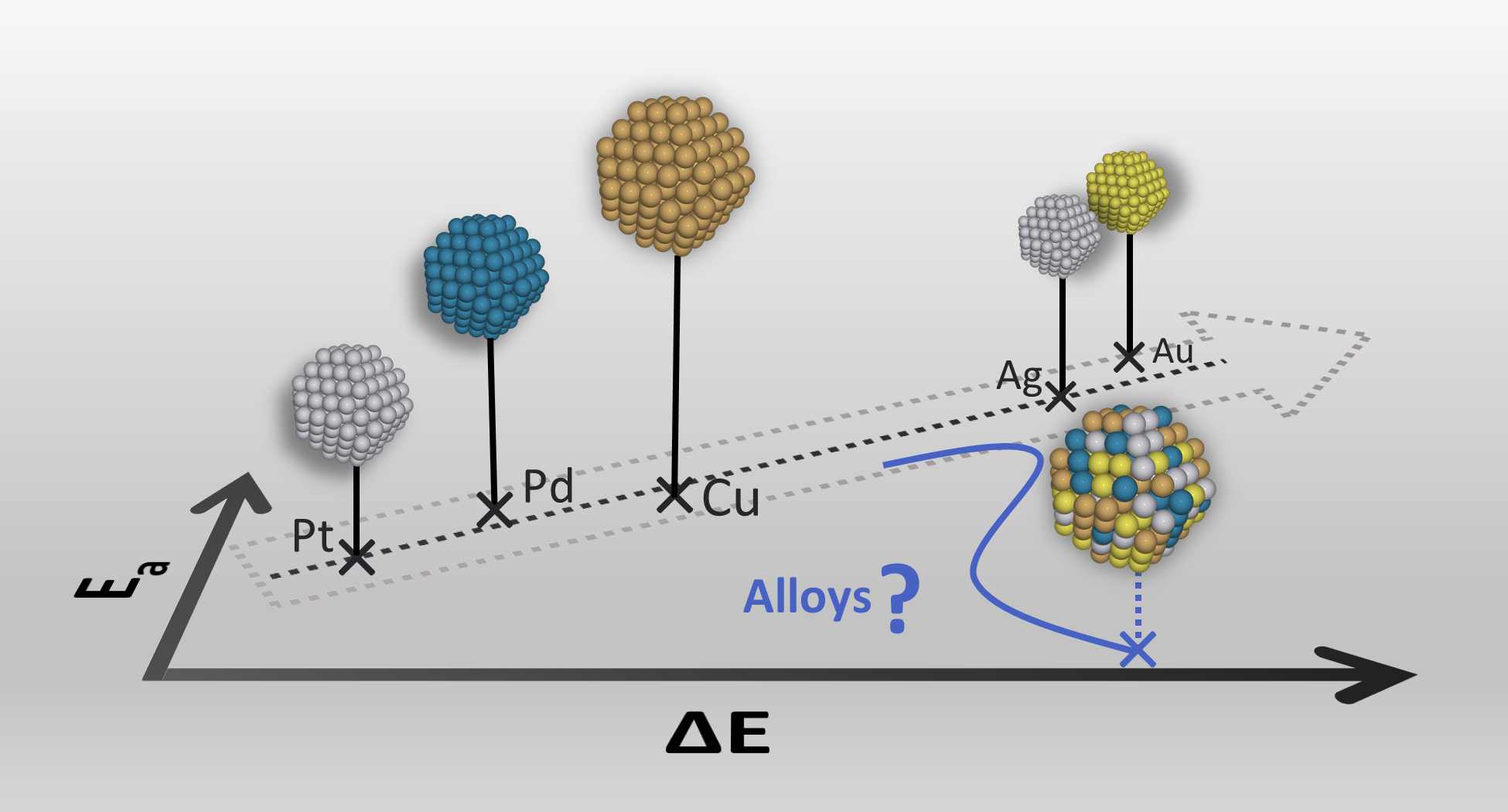
High-Entropy Alloys as a Discovery Platform for Electrocatalysis |
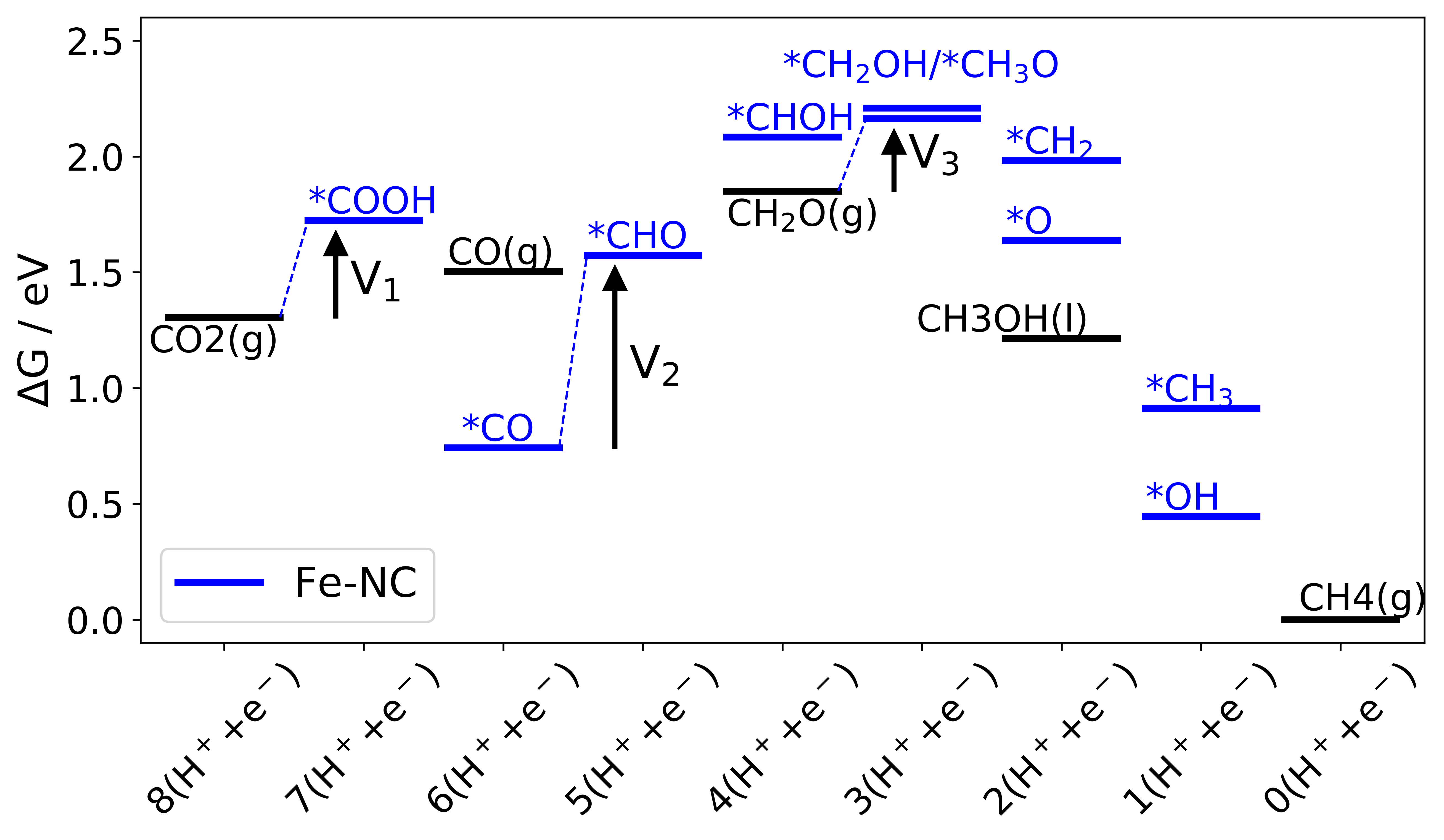 |
|
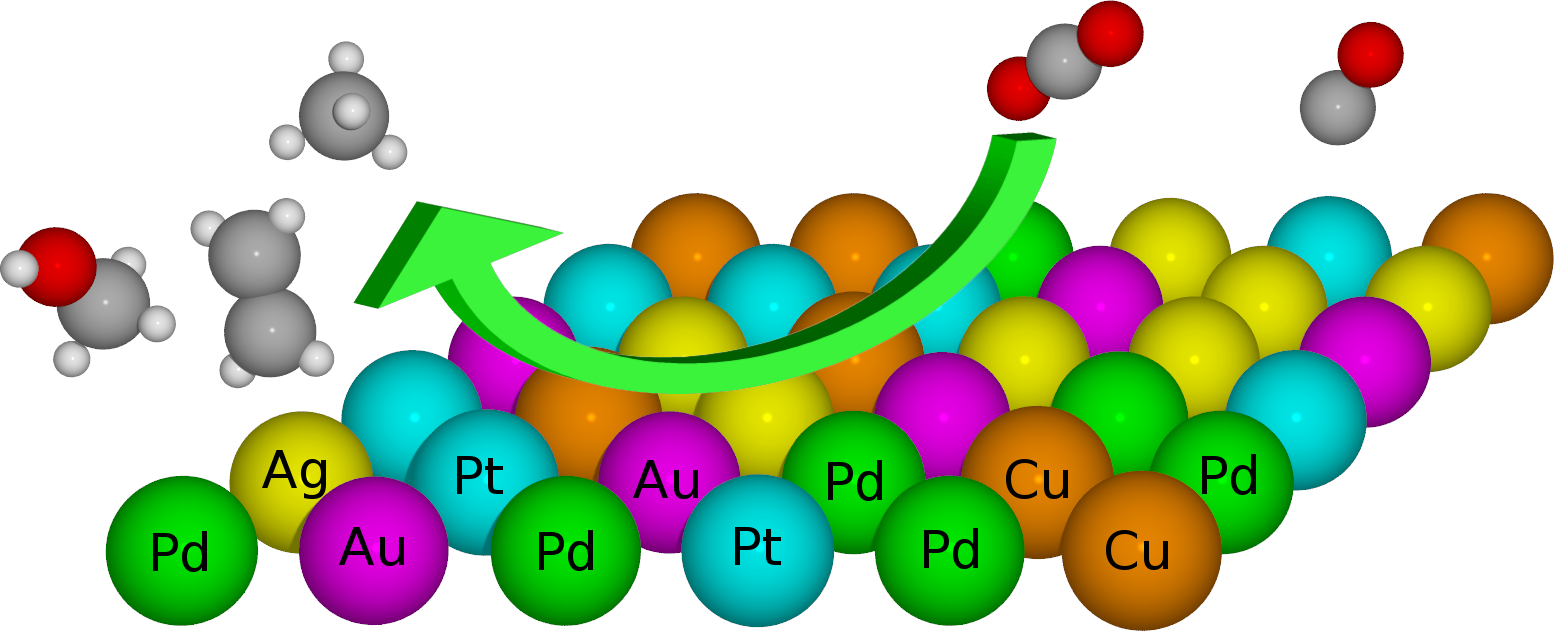 |
|
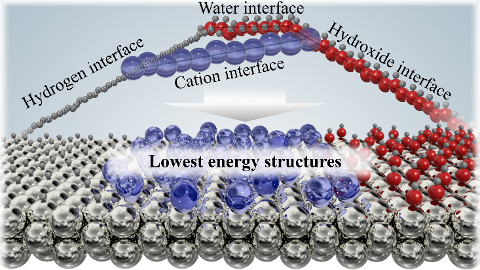 |
Realistic Cyclic Voltammograms from Ab Initio Simulations in Alkaline and Acidic Electrolytes Contains a .zip file with databases and scripts for plotting the figures from the paper. |
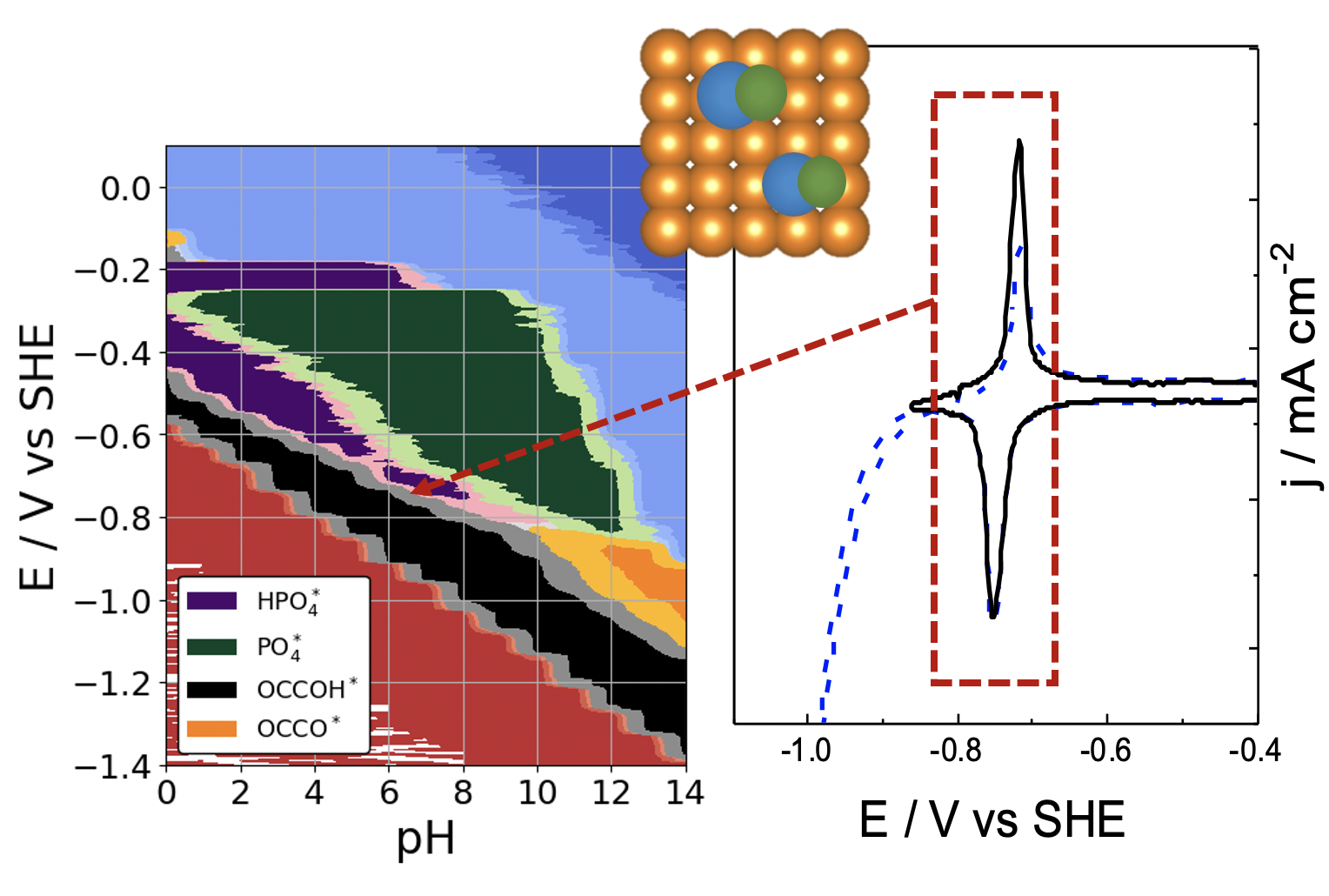 |
pH and anion effects on Cu-phosphate interfaces for CO electroreduction Contains a .zip file with databases and scripts for plotting the figures from the paper. |
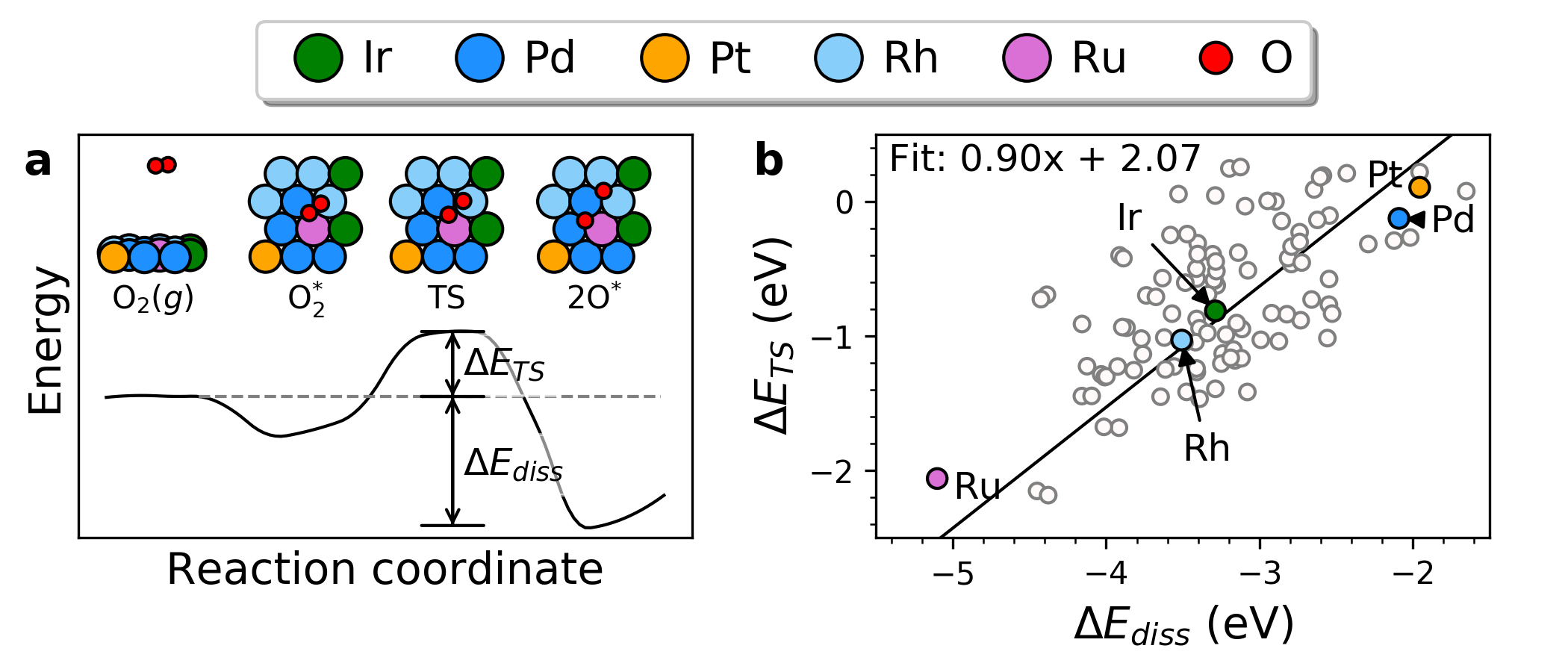 |
|
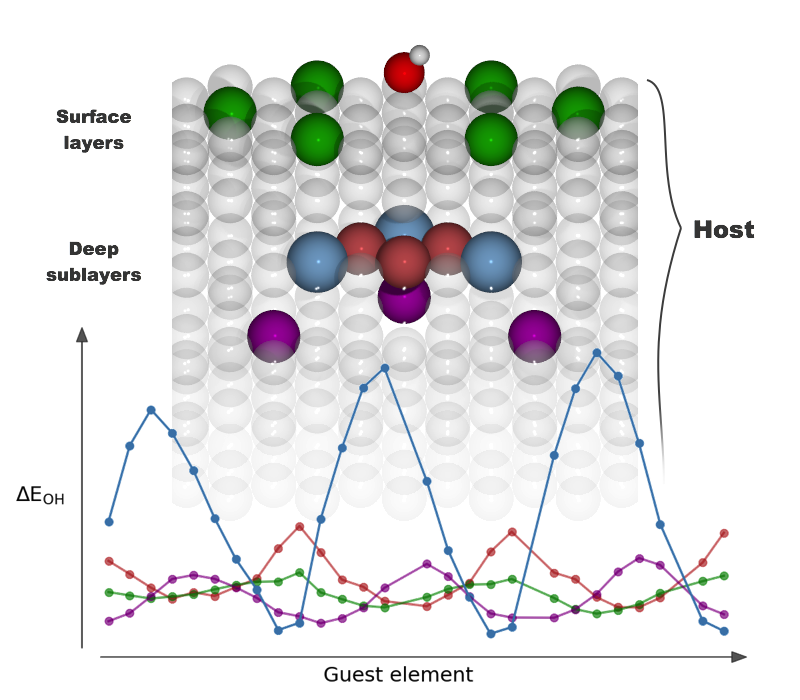 |
Long‐Range, Directional Ligand Effects in Metallic Alloys A statistical analysis uncovers what atomic positions, near the binding site on the surface of an alloy, exert an electronic effect on the adsorbate–surface bond. |
 |
Capturing the Formation Mechanism of Additive-Mediated Intermetallic PdCu Nanoparticles Files for reproducing the DFT calculations on the stability of the investigated phases of PdCu. |
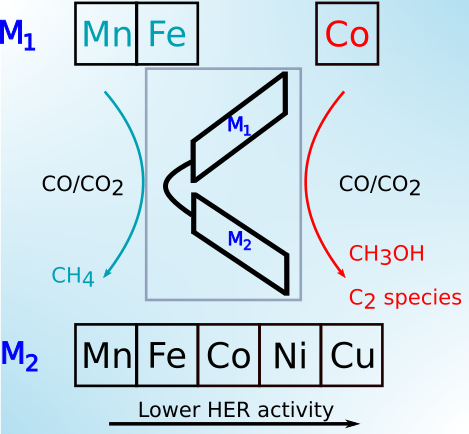 |
Three-Dimensional Carbon Electrocatalysts for CO2 or CO Reduction Contains a database file with structures and energies, and scrips for plotting all figures from the paper. |
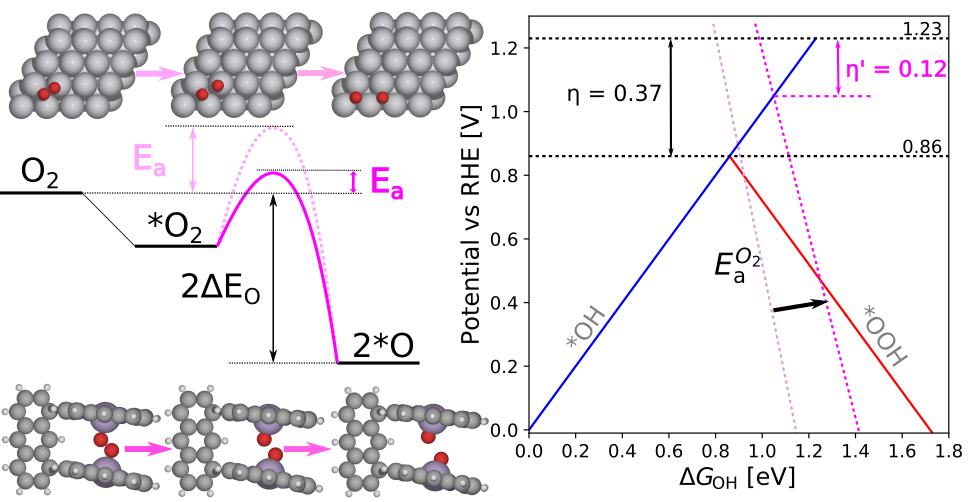 |
Insights in the Oxygen Reduction Reaction: From Metallic Electrocatalysts to Diporphyrins Contains a database file with all the structures for the paper. |
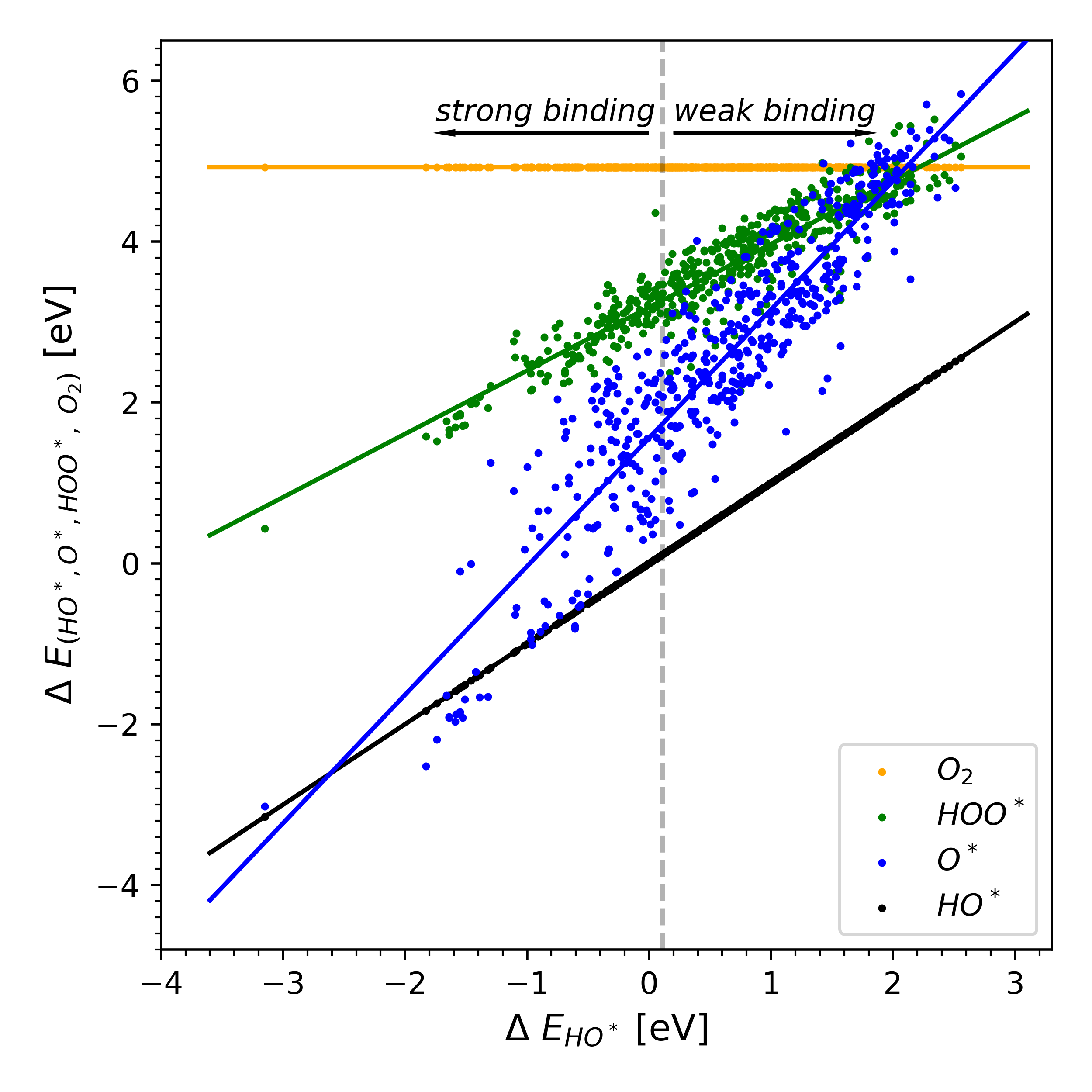 |
|
| Lattice distortion releasing local surface strain on high-entropy alloys
|
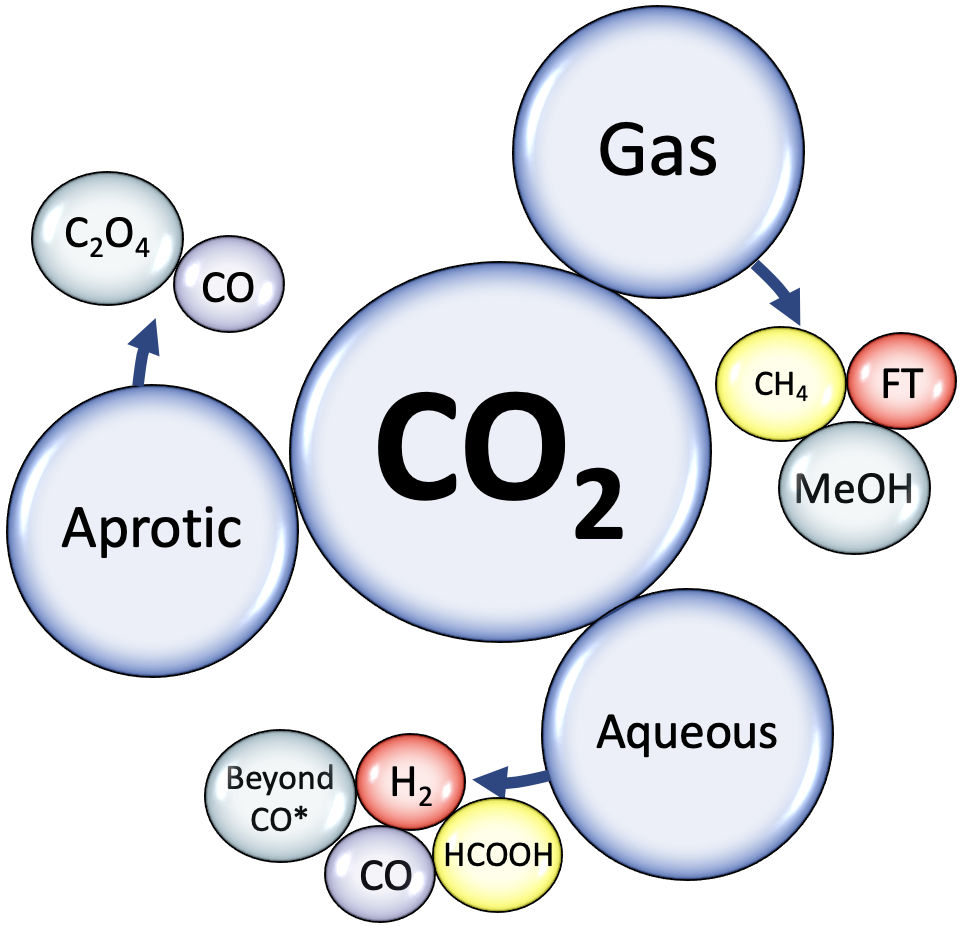
How to use the database: